Fe Mцssbauer Study of Unusual Magnetic Structure of Multiferroic 3R
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Superferromagnetism and Domain-Wall Topologies in Artificial
This is an open access article published under a Creative Commons Attribution (CC-BY) License, which permits unrestricted use, distribution and reproduction in any medium, provided the author and source are cited. Article Cite This: ACS Nano XXXX, XXX, XXX−XXX www.acsnano.org Superferromagnetism and Domain-Wall Topologies in Artificial “Pinwheel” Spin Ice † † † ‡ † Yue Li, Gary W. Paterson,*, Gavin M. Macauley, Fabio S. Nascimento, Ciaran Ferguson, ∇ § ∥ ∥ † † Sophie A. Morley, , Mark C. Rosamond, Edmund H. Linfield, Donald A. MacLaren, Rair Macedo,̂ ∇ † † ⊥ Christopher H. Marrows, Stephen McVitie,*, and Robert L. Stamps*, , † SUPA, School of Physics and Astronomy, University of Glasgow, Glasgow G12 8QQ, United Kingdom ‡ Departamento de Física, Universidade Federal de Vicosa,̧ Vicosa̧ 36570-900, Minas Gerais, Brazil ∇ School of Physics and Astronomy, University of Leeds, Leeds LS2 9JT, United Kingdom § Department of Physics, University of California, Santa Cruz, California 95064, United States ∥ School of Electronic and Electrical Engineering, University of Leeds, Leeds LS2 9JT, United Kingdom ⊥ Department of Physics and Astronomy, University of Manitoba, Manitoba R3T 2N2, Canada *S Supporting Information ABSTRACT: For over ten years, arrays of interacting single- domain nanomagnets, referred to as artificial spin ices, have been engineered with the aim to study frustration in model spin systems. Here, we use Fresnel imaging to study the reversal process in “pinwheel” artificial spin ice, a modified square ASI structure obtained by rotating each island by some angle about its midpoint. Our results demonstrate that a simple 45° rotation changes the magnetic ordering from antiferromagnetic to ferromagnetic, creating a superferromagnet which exhibits mesoscopic domain growth mediated by domain wall nucleation and coherent domain propagation. -

Final Program
1 General Information SCOPE OF THE CONFERENCE The 56th Magnetism and Magnetic Materials Conference is sponsored joint- ly by Physics Conferences Inc. and the Magnetics Society of the IEEE, in cooperation with the American Physical Society. Members of the interna- tional scientific and engineering communities interested in recent develop- ments in fundamental and applied magnetism are invited to attend the Con- ference and contribute to its technical sessions. Sessions will include invit- ed and contributed papers, oral and poster presentations, and invited symposia. This Conference provides an outstanding opportunity for world- wide participants to meet their colleagues and collaborators and discuss developments in all areas of magnetism research. In terms of the number of presentations, this will be the largest MMM in the history of the Conference, with over 1700 oral and poster presentations. PHOENIX/SCOTTSDALE, ARIZONA Scottsdale (www.ScottsdaleAz.gov/) is a suburb north of Phoenix located in the beautiful Sonoran Desert and bordered by the McDowell Mountains. The McDowell Sonoran Preserve is the largest urban wilderness areas in the U.S. The city of Scottsdale, which is historic in its own right, offers approx- imately 100 art galleries and 85 restaurants. TRANSPORTATION Phoenix Sky Harbor International Airport (PHX) (www.ifly.com/phoenix- sky-harbor-international-airport) is served by a host of airlines offering direct and connecting flights to all parts of the world. To reach the JW Mar- riott Desert Ridge Resort & Spa, where all MMM 2011 sessions and activ- ities will be held, there is a Shared Ride Van service available from “Super- Shuttle.” The current one-way cost in a shared ride van holding a total of 7 passengers is $22/person. -

Supermagnetism in Magnetic Nanoparticle Systems
Supermagnetism in magnetic nanoparticle systems (Supermagnetismus in magnetischen Nanoteilchensystemen) Vom Fachbereich Physik der Universität Duisburg-Essen (Campus Duisburg) zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigte Dissertation von Subhankar Bedanta aus Jignipur, Cuttack, Indien Referent : Prof. Dr. Wolfgang Kleemann Korreferent : Prof. Dr. Michael Farle Tag der mündlichen Prüfung : 11. Dezember 2006 Dedicated to my parents I Abstract Nanoscale magnetic materials are of interest for applications in ferrofluids, high-density magnetic storage, high-frequency electronics, high performance permanent magnets, and, magnetic refrigerants. Magnetic single-domain nanoparticles (“superspins) are very interesting not only for potential applications, e.g. high density storage devices, but also for fundamental research in magnetism. In an ensemble of nanoparticles in which the inter- particle magnetic interactions are sufficiently small, the system shows superparamagnetic (SPM) behavior as described by the Néel-Brown model. On the contrary, when inter- particle interactions are non-negligible, the system eventually shows collective behavior, which overcomes the individual anisotropy properties of the particles. In order to address the effect of interactions, we have investigated two different magnetic nanoparticle systems. The first part of this thesis focuses on the magnetic properties of ensembles of magnetic single-domain nanoparticles in an insulating matrix. The samples have a granular multilayer structure prepared as discontinuous metal-insulator multilayers (DMIM) [Co80Fe20 (tn)/Al2O3 (3nm)]m where the nominal thickness of CoFe is varied in the range 0.5 £ tn £ 1.8 nm, and the number of bilayers m is varied between 1- 10. The DMIMs represent a model system to study the effect of inter-particle interactions by varying the nominal thickness which corresponds to the magnetic particle concentration. -

237298246.Pdf
This is the submitted version of the following article: Roca A.G., Gutiérrez L., Gavilán H., Fortes Brollo M.E., Veintemillas-Verdaguer S., Morales M.D.P.. Design strategies for shape-controlled magnetic iron oxide nanoparticles. Advanced Drug Delivery Reviews, (2019). 138. : 68 - . 10.1016/j.addr.2018.12.008, which has been published in final form at https://dx.doi.org/10.1016/j.addr.2018.12.008 © https://dx.doi.org/10.1016/j.addr.2018.12.008. This manuscript version is made available under the CC-BY-NC-ND 4.0 license http://creativecommons.org/licenses/by-nc-nd/4.0/ Design Strategies for Shape-Controlled Magnetic Iron Oxide Nanoparticles Alejandro G. Rocaa,b,*, Lucía Gutiérreza,c,*, Helena Gavilána, María Eugênia Fortes Brolloa, Sabino Veintemillas-Verdaguera, María del Puerto Moralesa a, Instituto de Ciencia de Materiales de Madrid, Consejo Superior de Investigaciones Científicas, Cantoblanco, E-28049 Madrid, Spain b Catalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and BIST, Campus UAB, Bellaterra, E-08193 Barcelona, Spain cDept. Química Analítica, Instituto de Nanociencia de Aragón, Universidad de Zaragoza and CIBER-BBN, E-50018 Zaragoza, Spain Corresponding author: E-mail: [email protected]; [email protected] Keywords: Magnetic nanoparticles; anisometry; shape anisotropy; nanocubes; elongated nanoparticles; disks; hollow nanoparticles; nanocitotoxicity; biomedical applications Abstract: Ferrimagnetic iron oxide nanoparticles (magnetite or maghemite) have been the subject of an intense research since ancient times, not only for fundamental research but also for their potentiality in a widespread number of practical applications. Most of these studies were focused on nanoparticles with spherical morphology but recently there is an emerging interest on anisometric nanoparticles. -

Superparamagnetic Nanoparticle Ensembles
1 Superparamagnetic nanoparticle ensembles O. Petracic Institute of Experimental Physics/Condensed Matter Physics, Ruhr-University Bochum, 44780 Bochum, Germany Abstract: Magnetic single-domain nanoparticles constitute an important model system in magnetism. In particular ensembles of superparamagnetic nanoparticles can exhibit a rich variety of different behaviors depending on the inter-particle interactions. Starting from isolated single-domain ferro- or ferrimagnetic nanoparticles the magnetization behavior of both non-interacting and interacting particle-ensembles is reviewed. A particular focus is drawn onto the relaxation time of the system. In case of interacting nanoparticles the usual Néel-Brown relaxation law becomes modified. With increasing interactions modified superparamagnetism, spin glass behavior and superferromagnetism are encountered. 1. Introduction Nanomagnetism is a vivid and highly interesting topic of modern solid state magnetism and nanotechnology [1-4]. This is not only due to the ever increasing demand for miniaturization, but also due to novel phenomena and effects which appear only on the nanoscale. That is e.g. superparamagnetism, new types of magnetic domain walls and spin structures, coupling phenomena and interactions between electrical current and magnetism (magneto resistance and current-induced switching) [1-4]. In technology nanomagnetism has become a crucial commercial factor. Modern magnetic data storage builds on principles of nanomagnetism and this tendency will increase in future. Also other areas of nanomagnetism are commercially becoming more and more important, e.g. for sensors [5] or biomedical applications [6]. Many potential future applications are investigated, e.g. magneto-logic devices [7], [8], photonic systems [9, 10] or magnetic refrigeration [11, 12]. 2 In particular magnetic nanoparticles experience a still increasing attention, because they can serve as building blocks for e.g. -

Magnetization Dynamics and Related Phenomena in Nanostructures Sayan Chandra University of South Florida, [email protected]
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School January 2013 Magnetization Dynamics and Related Phenomena in Nanostructures Sayan Chandra University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the Condensed Matter Physics Commons, and the Nanoscience and Nanotechnology Commons Scholar Commons Citation Chandra, Sayan, "Magnetization Dynamics and Related Phenomena in Nanostructures" (2013). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/4877 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Magnetization Dynamics and Related Phenomena in Nanostructures by Sayan Chandra A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Physics College of Arts and Sciences University of South Florida Major Professor: Hariharan Srikanth, Ph.D. Pritish Mukherjee, Ph.D. Casey Miller, Ph.D. Inna Ponomareva, Ph.D. Manh-Huong Phan, Ph.D. Date of Approval: October 28, 2013 Keywords: Magnetism, Exchange Bias, Core/shell, Manganites, Spin glass Copyright © 2013, Sayan Chandra DEDICATION I dedicate this dissertation to my mother Mrs. Manju Chandra, my father Mr. Sushanta Kumar Chandra, my wife Mrs. Priyanka Dutta, and my sister Ms. Srijita Chandra for their constant support and faith in my abilities. I would also like to recognize my cousin Mr. Souvik Kumar Bari and my uncle Mr. Asis Kumar Das Kanungo for their help during my formative years. -

Collective States of Interacting Ferromagnetic Nanoparticles
ARTICLE IN PRESS Journal of Magnetism and Magnetic Materials 300 (2006) 192–197 www.elsevier.com/locate/jmmm Collective states of interacting ferromagnetic nanoparticles O. Petracica, X. Chena, S. Bedantaa, W. Kleemanna,Ã, S. Sahoob, S. Cardosoc, P.P. Freitasc aAngewandte Physik, Universita¨t Duisburg-Essen, D-47048 Duisburg, Germany bDepartment of Physics and Astronomy, University of Nebraska, Lincoln, Nebraska 68588, USA cINESC, Rua Alves Redol 9-1, 1000 Lisbon, Portugal Available online 15 November 2005 Abstract Discontinuous magnetic multilayers [CoFe/Al2O3] are studied by use of magnetometry, susceptometry and numeric simulations. Soft ferromagnetic Co80Fe20 nanoparticles are embedded in a diamagnetic insulating a-Al2O3 matrix and can be considered as homogeneously magnetized superspins exhibiting randomness of size (viz. moment), position and anisotropy. Lacking intra-particle core-surface ordering, generic freezing processes into collective states rather than individual particle blocking are encountered. With increasing particle density one observes first superspin glass and then superferromagnetic domain state behavior. The phase diagram resembles that of a dilute disordered ferromagnet. Criteria for the identification of the individual phases are given. r 2005 Elsevier B.V. All rights reserved. PACS: 75.10.Nr; 75.50.Lk; 75.40.Gb; 75.60.Ej Keywords: Magnetic nanoparticles; Dipolar interaction; Superspin glass; Superferromagnetism À10 1. Introduction where t010 s is the inverse attempt frequency, K an effective anisotropy constant and V the volume of the The physics of nanoscale magnetic materials is a vivid nanoparticle. The energy barrier is here approximated by subject in current magnetism research. This is partially due EB ¼ KV. to the promised potential in modern data storage applica- The magnetic behavior of the particle is characterized by tions [1] but mainly due to the wide spectrum of novel the so-called ‘‘blocking’’ temperature, Tb, below which the effects found [2–4]. -

Magnetic Interactions Between Nanoparticles
Magnetic interactions between nanoparticles Steen Mørup*1, Mikkel Fougt Hansen2 and Cathrine Frandsen1 Review Open Access Address: Beilstein J. Nanotechnol. 2010, 1, 182–190. 1Department of Physics, Building 307; Technical University of doi:10.3762/bjnano.1.22 Denmark; DK-2800 Kongens Lyngby; Denmark and 2Department of Micro- and Nanotechnology, DTU Nanotech, Building 345 East; Received: 10 September 2010 Technical University of Denmark; DK-2800 Kongens Lyngby; Accepted: 03 December 2010 Denmark Published: 28 December 2010 Email: Guest Editors: U. Wiedwald and P. Ziemann Steen Mørup* - [email protected] © 2010 Mørup et al; licensee Beilstein-Institut. * Corresponding author License and terms: see end of document. Keywords: dipole interactions; exchange interactions; spin structure; superferromagnetism; superparamagnetic relaxation Abstract We present a short overview of the influence of inter-particle interactions on the properties of magnetic nanoparticles. Strong magnetic dipole interactions between ferromagnetic or ferrimagnetic particles, that would be superparamagnetic if isolated, can result in a collective state of nanoparticles. This collective state has many similarities to spin-glasses. In samples of aggregated magnetic nanoparticles, exchange interactions are often important and this can also lead to a strong suppression of superparamag- netic relaxation. The temperature dependence of the order parameter in samples of strongly interacting hematite nanoparticles or goethite grains is well described by a simple mean field model. Exchange interactions between nanoparticles with different orientations of the easy axes can also result in a rotation of the sub-lattice magnetization directions. Review Introduction In nanostructured magnetic materials, interactions between, for hysteresis curves obtained after field cooling of a ferromag- example, nanoparticles or thin films in multilayer structures netic or ferrimagnetic material in contact with an antiferromag- often play an important role. -

Physics Reports Room Temperature Magnetism in Ceo2—A Review
Physics Reports ( ) – Contents lists available at ScienceDirect Physics Reports journal homepage: www.elsevier.com/locate/physrep Room temperature magnetism in CeO2—A review Karl Ackland, J.M.D. Coey * School of Physics and CRANN, Trinity College, Dublin 2, Ireland article info a b s t r a c t Article history: Cerium dioxide clearly raises the question of whether spontaneous ferromagnetic order is Accepted 9 April 2018 possible at high temperatures without d- electrons. There are many reports in the literature Available online xxxx of a ferromagnetic-like response to an applied magnetic field at room temperature for bulk, Editor: F. Parmigiani nanocrystalline or thin film samples, with or without cation doping. Typical values of the saturation magnetization are very small, of order 0.1 kAm−1, but reports range from zero Keywords: up to 1000 kAm−1. The effect is somehow related to lattice defects – Ce3C cations or oxygen Ferromagnetism d-zero magnetism vacancies – but it is a challenge to understand how electrons associated with these defects Exchange could order ferromagnetically at room temperature and above. Straightforward impurity Crystal defects effects are considered, and models based on conventional ferromagnetic superexchange Zero-point fluctuations or double exchange are discussed, as is exchange splitting of the 4f band or a defect- Giant orbital paramagnetism related impurity band. Results are also compared with a new model of athermal giant orbital paramagnetism that involves no spontaneous ferromagnetic order. A key issue is the fraction, if any, of the volume of the CeO2 samples that is spontaneously ferromagnetic. Detailed analysis of the magnetic properties suggests that the conventional explanations of the magnetism of CeO2 are untenable, and directions for further research are suggested. -
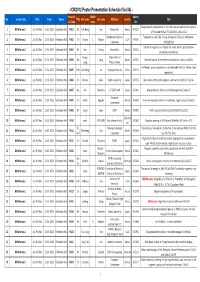
ICM2012 Session List 120702
- ICM2012 Poster Presentation Schedule (Vol.04) - Presentat Middle Abtract No. Session title Date Time Room Title First name Last name Affiliation Country Title ion Code name No. Magnetoelectric polarization in the field-induced commensurate phase 1 Multiferroics I July 9 (Mon) 13:30- 15:30 Exhibition Hall PA01 Mr. Hak Bong Lee Korea Univ. Korea AF0157 of Y-hexaferrite Ba0.7Sr1.3Zn2(Fe1-xAlx)12O22 Brookhaven National Temperature- and Field-Tuning of Magnetic Phases in Multiferroic 2 Multiferroics I July 9 (Mon) 13:30- 15:30 Exhibition Hall PA02 Dr. Christie S. Nelson USA AF0240 Laboratory NdFe3(BO3)4 Control of magnetic anisotropies for stable electric polarization in 3 Multiferroics I July 9 (Mon) 13:30- 15:30 Exhibition Hall PA03 Mr. Hun Chang Korea Univ. Korea AF0310 multiferroics hexaferrites Young- Department of 4 Multiferroics I July 9 (Mon) 13:30- 15:30 Exhibition Hall PA04 Mr. Song Korea AF0352 Chemical control of ferroelectric polarization in Mn1-xCoxWO4 Sang Physics, Korea Multiferroic phase competition in orthorhombic RMnO_3: Monte Carlo 5 Multiferroics I July 9 (Mon) 13:30- 15:30 Exhibition Hall PA05 Prof. Jun-Ming Liu Nanjing University China AF0437 approaches 6 Multiferroics I July 9 (Mon) 13:30- 15:30 Exhibition Hall PA06 Dr. Mitsuru Akaki Sophia University Japan AF0775 Spin-driven electric polarization in akermanite Sr2MSi2O7 crystals 7 Multiferroics I July 9 (Mon) 13:30- 15:30 Exhibition Hall PA07 Ms. Shin Miyahara JST ERATO-MF Japan AF0802 Magnetoelectric effects in antiferromagnet Ba2CoGe2O7 European 8 Multiferroics I July 9 (Mon) 13:30- 15:30 Exhibition Hall PA08 Dr. -

Magnetic Nanoparticles: a Subject for Both Fundamental Research and Applications
Hindawi Publishing Corporation Journal of Nanomaterials Volume 2013, Article ID 952540, 22 pages http://dx.doi.org/10.1155/2013/952540 Review Article Magnetic Nanoparticles: A Subject for Both Fundamental Research and Applications S. Bedanta,1 A. Barman,2 W. Kleemann,3 O. Petracic,4 and T. Seki5 1 School of Physical Sciences, National Institute of Science Education and Research (NISER), IOP Campus, Bhubaneswar 751005, India 2 Department of Condensed Matter Physics and Material Sciences, S. N. Bose National Centre for Basic Sciences, Block-JD, Sector-III, Salt Lake City, Kolkata 700098, India 3 Department of Physics, University Duisburg-Essen, 47057 Duisburg, Germany 4 Julich¨ Centre for Neutron Science JCNS and Peter Grunberg¨ Institute PGI, JARA-FIT, Forschungszentrum Julich¨ GmbH, 52425 Julich,¨ Germany 5 Institute for Materials Research, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan Correspondence should be addressed to S. Bedanta; [email protected] Received 16 July 2013; Accepted 19 October 2013 Academic Editor: Gaurav Mago Copyright © 2013 S. Bedanta et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Single domain magnetic nanoparticles (MNPs) have been a vivid subject of intense research for the last fifty years. Preparation of magnetic nanoparticles and nanostructures has been achieved by both bottom-up and top-down approaches. Single domain MNPs show Neel-Brown-like´ relaxation. The Stoner-Wohlfarth model describes the angular dependence of the switching ofthe magnetization of a single domain particle in applied magnetic fields. -

Superferromagnetism and Domain-Wall Topologies in Artificial “Pinwheel” Spin
This is an open access article published under a Creative Commons Attribution (CC-BY) License, which permits unrestricted use, distribution and reproduction in any medium, provided the author and source are cited. Article Cite This: ACS Nano 2019, 13, 2213−2222 www.acsnano.org Superferromagnetism and Domain-Wall Topologies in Artificial “Pinwheel” Spin Ice † † † ‡ † Yue Li, Gary W. Paterson,*, Gavin M. Macauley, Fabio S. Nascimento, Ciaran Ferguson, ∇ § ∥ ∥ † † Sophie A. Morley, , Mark C. Rosamond, Edmund H. Linfield, Donald A. MacLaren, Rair Macedo,̂ ∇ † † ⊥ Christopher H. Marrows, Stephen McVitie,*, and Robert L. Stamps*, , † SUPA, School of Physics and Astronomy, University of Glasgow, Glasgow G12 8QQ, United Kingdom ‡ Departamento de Física, Universidade Federal de Vicosa,̧ Vicosa̧ 36570-900, Minas Gerais, Brazil ∇ School of Physics and Astronomy, University of Leeds, Leeds LS2 9JT, United Kingdom § Department of Physics, University of California, Santa Cruz, California 95064, United States ∥ School of Electronic and Electrical Engineering, University of Leeds, Leeds LS2 9JT, United Kingdom ⊥ Department of Physics and Astronomy, University of Manitoba, Manitoba R3T 2N2, Canada *S Supporting Information ABSTRACT: For over ten years, arrays of interacting single- domain nanomagnets, referred to as artificial spin ices, have been engineered with the aim to study frustration in model spin systems. Here, we use Fresnel imaging to study the reversal process in “pinwheel” artificial spin ice, a modified square ASI structure obtained by rotating each island by some angle about its midpoint. Our results demonstrate that a simple 45° rotation changes the magnetic ordering from antiferromagnetic to ferromagnetic, creating a superferromagnet which exhibits mesoscopic domain growth mediated by domain wall nucleation and coherent domain propagation.