CH 2270/2290 Extraction: a Separation and Isolation Technique (Adapted from Laboratory Manual to Accompany Organic Chemistry: a Short Course, H
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

P-Chloro-O-Toluidine and Its Hydrochloride Al
Report on Carcinogens, Fourteenth Edition For Table of Contents, see home page: http://ntp.niehs.nih.gov/go/roc p-Chloro-o-toluidine and Its Hydrochloride al. 1979, Bentley et al. 1986, IARC 2000). In organs from animals ex- posed to p-chloro-o-toluidine, DNA breakage was detected by single- CAS Nos. 95-69-2 and 3165-93-3 cell gel electrophoresis (comet assay) in mouse liver, urinary bladder, lung, and brain and in rat liver and kidney (Sekihashi et al. 2002). Reasonably anticipated to be human carcinogens First listed in the Eighth Report on Carcinogens (1998) Properties Also known as 4-chloro-o-toluidine or 4-chloro-2-methylaniline p-Chloro-o-toluidine is a chlorinated aromatic amine that exists as a grayish-white crystalline solid or leaflet, andp -chloro-o-toluidine hy- CH3 drochloride is a buff-colored or light-pink powder at room tempera- ture. The base compound is practically insoluble in water or carbon NH2 tetrachloride but is soluble in ethanol or dilute acid solutions. It is stable under normal temperatures and pressures (Akron 2009). Phys- Cl ical and chemical properties of p-chloro-o-toluidine are listed in the following table. No physical and chemical properties for the hydro- Carcinogenicity chloride were found except its molecular weight of 178.1 and melt- p-Chloro-o-toluidine and its hydrochloride salt are reasonably antic‑ ing range of 265°C to 270°C (IARC 2000, Weisburger 1978). ipated to be human carcinogens based on limited evidence of carci- Property Information nogenicity from studies in humans and evidence of carcinogenicity Molecular weight 141.6a from studies in experimental animals. -

PDMS-Based Antimicrobial Surfaces for Healthcare Applications
PDMS-based Antimicrobial Surfaces for Healthcare Applications This thesis is presented to UCL in partial fulfilment of the requirements for the Degree of Doctor of Philosophy Ekrem Ozkan Supervised by Professor Ivan P. Parkin Materials Chemistry Research Centre Dr Elaine Allan Division of Microbial Diseases 2017 1 DECLARATION I, Ekrem Ozkan confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 ABSTRACT This thesis describes two types of approaches for reducing the incidence of hospital- acquired infections (HAIs), which are chemical approaches that inactivate bacteria that adhere to the surface i.e. bactericidal activity and physical approaches that inhibit initial bacterial attachment to the surface i.e. anti-biofouling activity. Specifically, the antimicrobial polydimethylsiloxane (PDMS)-based systems detailed in this thesis are: (i) photosensitizer, crystal violet (CV),-coated PDMS for both medical device and hospital touch surface applications, (ii) crystal violet-coated, zinc oxide nanoparticle-encapsulated PDMS for hospital touch surface applications, (iii) superhydrophobic antibacterial copper coated PDMS films via aerosol assisted chemical vapour deposition (AACVD) for hospital touch surface applications and (iv) slippery copper-coated PDMS films to prevent biofilm formation on medical devices. The materials were characterized using techniques including: X-ray diffraction (XRD), scanning electron microscopy (SEM), UV-vis absorbance spectroscopy, water-contact angle measurement and microbiology tests. Functional testing indicated that CV-coated samples were suitable for targeted applications and showed potent light-activated antimicrobial activity when tested against model Gram-positive bacteria, Staphylococcus aureus, and Gram-negative bacteria, Escherichia coli, associated with hospital-acquired infections, with > 4 log reduction in viable bacterial numbers observed. -

Ortho-TOLUIDINE
ortho-TOLUIDINE 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Nomenclature Chem. Abstr. Serv. Reg. No.: 95–53–4 CAS Name: 2-Methylbenzenamine Synonyms: 1-Amino-2-methylbenzene; 2-amino-1-methylbenzene; 2-aminotoluene; ortho-aminotoluene; 2-methyl-1-aminobenzene; 2-methylaniline; ortho-methylaniline; ortho-methylbenzenamine; 2-methylphenylamine; 2- tolylamine; ortho-tolylamine 1.1.2 Structural formula, molecular formula, and relative molecular mass NH2 CH3 C7H9N Rel. mol. mass: 107.15 1.1.3 Chemical and physical properties of the pure substance Description: Light yellow liquid becoming reddish brown on exposure to air and light (O’Neil, 2006) Boiling-point: 200–201 °C (O’Neil, 2006) Melting-point: −14.41 °C (Lide, 2008) Solubility: Slightly soluble in water; soluble in diethyl ether, ethanol and dilute acids (O’Neil, 2006) Octanol/water partition coefficient: log P, 1.29–1.32 (Verschueren, 2001) –407– 408 IARC MONOGRAPHS VOLUME 99 1.1.4 Technical products and impurities No information was available to the Working Group. 1.1.5 Analysis ortho-Toluidine is the most widely studied of the aromatic amines reviewed in this Volume. Waste-water, air and urine samples have been analysed. The use of LC/MS and LC with electrochemical detection provides detection limits down to the nanomol level. ortho-Toluidine is one of the amines detected as a reduction product of azo dyes used as toy colourants. Table 1.1 presents selected methods of detection and quantification of ortho-toluidine in various matrices. Table 1.1. Selected methods of analysis of ortho-toluidine in various matrices Sample Sample preparation Assay method Detection limit Reference matrix Toys Sodium dithionite HPLC/UV 0.2 μg/g Garrigós et al. -

EXPERIMENT 2 SEPARATION of a MIXTURE of P-TOLUIDINE AND
EXPERIMENT 2 1:: SEPARATION OF A MIXTURE OF p-TOLUIDINE AND NAPHTHALENE BY SOLVENT EXTRACTION AND IDENTI- FICATION OF THEIR FUNCTIONAL GROUPS 4: 2.1 Introduction Objectives - 2.2 Principle 2.3 Requirements Identification of functional.groups 2.5 Result 2.1 INTROD-UCTION In the previous experiment you have separated a mixture containing two acidic and one neutral compound. The extracting solutions were aqueous bases. In this experiment you will be seperating a mixture of a basic and a neutral compound. As you will recall from sec. 1.2 of experiment 1 that this will require an aqueous'acidic solution. The basic compound after separation is recovered by re extraction. Objeetives After srudying and performing this experiment you should be able to : . separate a mixture of a neutral and a basic organic compound by solvent extraction technique, identify the functional group of the separated compounds, and describe the theory behind the above. 2.2 PRINCIPLE This mixture contains a basic compound, p-toluidine, and a neutral compound viz., naphthalene. Their separation is quite straight forward. You will recall from above that a basic compound can be extracted by an aqueous solution of an acid. As this mixture contains only one basic compound (as against two acidic compounds in the previous experiment), you don't need to bother about which acid to use for extraction. A strong acid like HCI is definitely a good choice and will afford an effective separation. The hydrochloric acid will convertp-toluidine into its hydrochloride salt which on extraction will moFVeinto aqueous layer. Separation of a Mixture of p-Toluidine and Naphthakne by Solvent Extraction and IdenH- Ucatkm dthelr FuaEtiooal C~P Q NII: HCI NI I,C'I- - + NH2 @-toluidine) @-tohidine hydrochldride) p-Toluidine Freep-toluidine can be recovered by hydrolping this salt with an aqueous base. -

Perkin's Mauve: the History of the Chemistry
REFLECTIONS Perkin’s Mauve: The History of the Chemistry Andrew Filarowski Those of us who owe our living in part to the global dyestuff and chemical industry should pause today and remember the beginnings of this giant industry which started 150 years ago today with William Perkins’ discovery of mauveine whilst working in his home laboratory during the Easter holiday on April 28, 1856. Prior to this discovery, all textiles were dyed with natural dyestuffs and pigments. What did Perkin’s Reaction Entail? William Henry Perkin carried out his experiments at his home laboratory in the Easter break of 1856. He was trying to produce quinine (C20H24N2O2). This formula was known but not the structural formula. Because chemistry was in such an early stage of development Perkin thought that by simply balancing the masses (simple additive and subtractive chemistry) in an equation he would obtain the required compound. He therefore believed that if he took two allyltoluidine molecules, C10H13N, and oxidised them with three oxygen atoms (using potassium dichromate) he would get quinine (C20H24N2O2) and water. 2 (C10H13N) + 3O C20H24N2O2 + H2O It is unsurprising to us now but Perkin reported “that no quinine was formed, but only a dirty reddish brown precipitate.” However, he continued in his trials and decided to use aniline (C6H5NH2) and its sulphate, and to oxidise them using potassium dichromate. This produced a black precipitate that Perkin at first took to be a failed experiment, but he noticed on cleaning his equipment with alcohol that a coloured solution was obtained. Perkin’s Patent W H Perkin filed his patent on the 26th August 1856 for “Producing a new colouring matter for the dyeing with a lilac or purple color stuffs of silk, cotton, wool, or other materials.” (sic) Patent No. -

Biosynthesis of Quinine and Related Alkaloids EDM-ARI)LEETE
February 1060 I~IOSYNTHESISOF QUINIXE AND l1ELATE:L) XLKALO1I)S 50 Biosynthesis of Quinine and Related Alkaloids EDM-ARI)LEETE Department oj Chenaisli.y, Ilniversitij of Minnesota, ilfinneapoIis, I1mnesotu 6,i/+ R~eizpdSeptendw ,I, 1968 The antimalarial drug quinine (1) has had a fascinat- years old at the timc of hwvestirig for quinirie extr:ic- ing history.’ In 1820 it was isolated in a crystalline tion. state from the bark of a Cinchona tree by l’elletier T’ortunately from our point of view, cjuiniiic mid and Caventou.2 In 1856 I’erltin, having little infor- other alkaloids are biosynthe*ized in young seedlings. mation on the alkaloid except its molecular formula, This was evident from the work of de Jloerloo~,~ attempted to obtain it by oxidizing allyltoluidine with who cultivated l-year-old Czr~chomsuccii-ubi~ p1:Lrit.: potassium dichromate according to in a11 atmosphere containing radioactive carbon dioxide and obtained radioactive quinine from the p1:mts. 2CioHiBN + 30 + C+NzO, + HzO allyltolkiidiiie yiiinine The biogenetic relationship of the indole illkdoids cin- Seedless to say, this attempt was unsuccessful and choriamine and quiriamine was suggested by Chuttirel, yielded a dirty reddish brown precipitate. However, et UZ.,~ and a plausible biogenetic scheme based on this the results of this experiment led Perkin to investigate view, upon Turner and Wood\vitrd’sg ideaq, arid oil the oxidation of simpler amines, and from the oxidation tracer work to be described is illustrated in Scheme I. of crude aniline (which contained some toluidine) he It is suggested that tryptophan (or possibly trypt- obtained the first synthetic dye, mauve (2),which was amine’O) condenses with the nine-carbon triuldehyde used for dyeing silk and coloring postage stamps in the 3 (the origin of this unit will be discussed later) to 186O’~.~ afford the tetrahydro-@-carbolinederivative 4. -

Separation and Identification of Aromatic Amines by Two
368 Z. Anal. Chem., Band 267, Heft 5 (1973) Separation and Identification Table 1. R~-Values of aromatic amines with n-heptane, of Aromatic Amines by Two-Dimensional chloroform and carbon tetraehloride Thin-Layer Chromatography S.No. Amines R~-values a (a) (b) (c) of Charge-Transfer Complexes Liquid amines Trennung und Identifizierung aromatischer Amine 1. Aniline 0.096 -- 0.37 durch zweidimensionale Dfinnschicht-Chromatographic 2. m-Toluidine 0.17 -- -- yon Charge-Transfer-Komplexen 3. o-Toluidine 0.19 -- -- 4. N-lYIethyl aniline 0.26 -- 0.73 Trenn. yon Aminen, aromatische; Chromatographic, 5. ~,N-Dimethyl aniline 0.46 ~ 0.89 Diinnsehieht; Charge-Transfer -Komplexe Solid amines J. P. Sharma and Sushma Ahuja 6. p-Aminobenzoic acid -- 0.03 -- Dept. of Chemistry, University Allahabad, India 7. Benzidine -- 0.09 -- Received March 9, 1973; revised l~ay 29, 1973 8. o-Aminobenzoie acid -- 0.12 -- 9. a-Naphthylamine -- 0.30 0.24 10. fl-Naphthylamine -- 0.30 0.17 Aromatic amines form weak association complexes 11. p-Toluidine -- 0.39 0.43 with aromatic nitro compounds and the association a--c ~ Solvents, see text. constants of the complexes formed with a given a All the Rf-values are average of three to four runs. aeceptor are usually different depending upon the donor strengths of amines. When m-dinitrobenzene inserted in the tanks which had already been saturated is impregnated in the thin-layers made of silica gel with solvent vapour for 10 rain. and the aromatic amines ehromatographed on these The plates were developed for 30--35 rain except in the layers, good separation is achieved. -

Synthesis, Characterization and Thermal Degradation Studies of O-Toluidine-Biuret-Formaldehyde Copolymer
International Journal of Multidisciplinary and Current Educational Research (IJMCER) ISSN: 2581-7027 ||Volume|| 3 ||Issue|| 3 ||Pages 346-352 ||2021|| Synthesis, Characterization and Thermal Degradation Studies of O-Toluidine-Biuret-Formaldehyde Copolymer Dinesh D. Kamdi Department of Science, Manawata Vidyalaya, Babulkheda, Nagpur-440027, Maharashtra, India. ABSTRACT: The copolymer resin o-Toluidine-Biuret-Formaldehyde (o-TBF) was synthesized by the condensation of o-toluidine and biuret with formaldehyde in the presence of 2M HCl as a catalyst at 124 ± 2 0C for 5 hrs with molar proportion of reactants. Thermogravimetric analysis of o-TBF copolymer has been performed to study the thermal degradation characteristics and to ascertain its thermal stability. In this technique the sample of o-TBF copolymer resin was subjected to the condition of continuous increase in temperature at linear rate.The thermal study of o-TBF copolymer was carried out to study it’s mode of decomposition and relative thermal stability. The copolymer resin o-TBF was characterized by UV-visible absorption spectra, FT-IR and proton NMR specteral studies. The thermal activation energy (Ea) calculated using above mentioned methods was in good agreement. The Freeman-Carroll and Sharp-Wentworth methods were used to calculate thermal activation energy (Ea), order of reaction (n), frequency factor (Z), entropy change (ΔS), free energy change (ΔF), and apparent entropy change (S*). The thermal activation energy (Ea) determined with these methods was in good agreement with each other. KEYWORDS: Synthesis, condensation, thermogravimetric analysis, decomposition, spectral studies, activation energy, kinetic parameters. I. INTRODUCTION Due to global applications of polymeric materials, polymer science and technology have been developing rapidly and attracted much attention towards the polymer scientists. -

Ortho-TOLUIDINE Ortho-Toluidine Was Considered by Previous IARC Working Groups in 1977, 1981, 1987, 2000, and 2008 (IARC, 1978, 1982, 1987, 2000, 2010)
ortho-TOLUIDINE ortho-Toluidine was considered by previous IARC Working Groups in 1977, 1981, 1987, 2000, and 2008 (IARC, 1978, 1982, 1987, 2000, 2010). Since that time new data have become avail- able, which have been incorporated in this Monograph, and taken into consideration in the present evaluation. 1. Exposure Data dyestuffs, azo pigment dyes, triarylmethane dyes, sulfur dyes, and indigo compounds), and 1.1 Identification of the agent as an intermediate for synthetic rubber and rubber-vulcanizing chemicals, pharmaceuticals, From IARC (2010), unless indicated otherwise pesticides, and other chemicals. ortho-Toluidine Chem. Abstr. Serv. Reg. No.: 95-53-4 is also used in the clinical laboratory as an ingre- Chem. Abstr. Serv. Name: dient in a reagent for glucose analysis, and for 2-Methylbenzenamine tissue staining (IARC, 2010; NTP, 2004). CH3 NH2 1.3 Human exposure 1.3.1 Occupational exposure Occupational exposure to ortho-toluidine C H N 7 9 can occur by inhalation or skin contact during Relative molecular mass: 107.15 its production, or during the production of dyes, Description: Light yellow liquid becoming pigments and rubber chemicals manufactured reddish brown on exposure to air and light from this chemical. Laboratory and medical Boiling-point: 200–202 °C (O’Neil, 2006) personnel may be exposed when using ortho- Solubility: Slightly soluble in water; soluble toluidine for staining tissues (IARC, 2010). in alcohol, ether, and dilute acids From the US National Occupational Exposure Survey (1981–83) it was estimated that 30000 1.2 Uses workers, including approximately 15500 women, were potentially exposed to ortho-toluidine ortho-Toluidine is used as an intermediate (NIOSH, 1990). -

P-Toluidine CAS N°: 106-49-0
OECD SIDS p-TOLUIDINE FOREWORD INTRODUCTION p-Toluidine CAS N°: 106-49-0 UNEP PUBLICATIONS 1 OECD SIDS p-TOLUIDINE SIDS Initial Assessment Report For SIAM 21 Washington, DC, 18–21 October 2005 1. Chemical Name: p-Toluidine 2. CAS Number: 106-49-0 3. Sponsor Country: Germany Contact Point: BMU (Bundesministerium für Umwelt, Naturschutz und Reaktorsicherheit) Contact person: Prof. Dr. Ulrich Schlottmann Postfach 12 06 29 D- 53048 Bonn 4. Shared Partnership with: 5. Roles/Responsibilities of the Partners: • Name of industry sponsor Bayer AG, Germany /consortium Contact person: Dr. Burkhardt Stock D-51368 Leverkusen Building 9115 • Process used The BUA Peer Review Process : see next page 6. Sponsorship History • How was the chemical or by ICCA-Initiative category brought into the OECD HPV Chemicals Programme? 7. Review Process Prior to last literature search (update): the SIAM: 14 April 2004 (Human Health): databases medline, toxline; search profile CAS-No. and special search terms 26 March 2004 (Ecotoxicology): databases CA, biosis; search profile CAS-No. and special search terms OECD/ICCA 8. Quality check process: IUCLID was used as a basis for the SIDS dossier. All data were checked and validated by BUA. A final evaluation of the human health part has been performed by the Federal Institute for Risk Assessment (BfR) and of the ecotoxicological part by the Federal Environment Agency (UBA). 9. Date of Submission: Deadline for circulation: 22 July 2005 10. Date of last Update: Last literature search: IUCLID Chapters 1-4: 2003-08-26, Chapter 5: 2004-01-02 2 UNEP PUBLICATIONS OECD SIDS p-TOLUIDINE 11. -

Ortho-TOLUIDINE
ortho-TOLUIDINE This substance was considered by previous working groups, in June 1977 (IARC, 1978), February 1981 (IARC, 1982) and March 1987 (IARC, 1987a). Since that time, new data have become available, and these have been incorporated in the monograph and taken into consideration in the evaluation. 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Nomenclature ortho-Toluidine Chem. Abstr. Serv. Reg. No.: 95-53-4 Chem. Abstr. Name: 2-Methylbenzenamine IUPAC Systematic Name: ortho-Toluidine Synonyms: 1-Amino-2-methylbenzene; 2-amino-1-methylbenzene; 2-amino- toluene; ortho-aminotoluene; 2-methyl-1-aminobenzene; 2-methylaniline; ortho- methylaniline; ortho-methylbenzenamine; 2-methylphenylamine; ortho-tolylamine ortho-Toluidine hydrochloride Chem. Abstr. Serv. Reg. No.: 636-21-5 Chem. Abstr. Name: 2-Methylbenzenamine hydrochloride IUPAC Systematic Name: ortho-Toluidine hydrochloride Synonyms: 1-Amino-2-methylbenzene hydrochloride; 2-amino-1-methylbenzene hydrochloride; 2-aminotoluene hydrochloride; ortho-aminotoluene hydrochloride; 2-methyl-1-aminobenzene hydrochloride; 2-methylaniline hydrochloride; ortho- methylaniline hydrochloride; ortho-methylbenzenamine hydrochloride; 2-methyl- phenylamine hydrochloride; ortho-tolylamine hydrochloride –267– 268 IARC MONOGRAPHS VOLUME 77 1.1.2 Structural and molecular formulae and relative molecular mass ortho-Toluidine NH2 CH3 C7H9N Relative molecular mass: 107.16 ortho-Toluidine hydrochloride NH2 CH3 HCl C7H9N.HCl Relative molecular mass: 143.62 1.1.3 Chemical and physical properties of the -
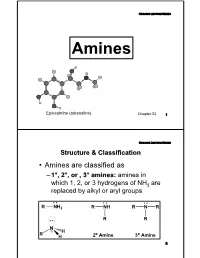
Basicity-Aromatic Amines
Organic Lecture Series AminesAmines Epinephrine (adrenaline) Chapter 23 1 Organic Lecture Series Structure & Classification • Amines are classified as – 1°, 2°, or , 3° amines: amines in which 1, 2, or 3 hydrogens of NH3 are replaced by alkyl or aryl groups 2 o o 2 Structure & ClassificationOrganic Lecture Series • Amines are further divided into aliphatic, aromatic, and heterocyclic amines: – aliphatic amine: an amine in which nitrogen is bonded only to alkyl groups – aromatic amine: an amine in which nitrogen is bonded to one or more aryl groups CH CH : 3 3 NH 2 N-H: CH2 -N-CH: 3 Aniline N-Methylaniline Benzyldimethylamine (a 1° aromatic amine) (a 2° aromatic amine) (a 3° aliphatic amine) 3 Organic Lecture Series Structure & Classification – heterocyclic amine: an amine in which nitrogen is one of the atoms of a ring N N N N H H H PyrrolidinePiperidine Pyrrole Pyridine (heterocyclic aliphatic amines) (heterocyclic aromatic amines) 4 Organic Lecture Series Structure & Classification CH3 N H H COOCH3 (a) (b) N (c) N CH OCH H N 3 6 5 (S)-Coniine (S)-Nicotine Co cain e O Coniine is a poisonous alkaloid found in poison hemlock and the Yellow Pitcher Plant, and contributes to hemlock's fetid smell. It is a neurotoxin which disrupts the peripheral nervous system. Caffeine 5 Organic Lecture Series Nomenclature • Aliphatic amines: replace the suffix -eofe the parent alkane by -amine NH2 NH2 H2 N NH2 2-Propanamine (S)-1-Phenyl- 1,6-Hexanediamine ethanamine 6 Organic Lecture Series Nomenclature • The IUPAC system retains the name aniline: NH2