The Role of the Transcription Factor SOX2 in Tumorigenesis and Development of the Stomach
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lower Genomic Stability of Induced Pluripotent Stem Cells Reflects
Zhang et al. Cancer Commun (2018) 38:49 https://doi.org/10.1186/s40880-018-0313-0 Cancer Communications ORIGINAL ARTICLE Open Access Lower genomic stability of induced pluripotent stem cells refects increased non‑homologous end joining Minjie Zhang1,2†, Liu Wang3†, Ke An1,2†, Jun Cai1, Guochao Li1,2, Caiyun Yang1, Huixian Liu1, Fengxia Du1, Xiao Han1,2, Zilong Zhang1,2, Zitong Zhao1,2, Duanqing Pei4, Yuan Long5, Xin Xie5, Qi Zhou3 and Yingli Sun1* Abstract Background: Induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs) share many common features, including similar morphology, gene expression and in vitro diferentiation profles. However, genomic stability is much lower in iPSCs than in ESCs. In the current study, we examined whether changes in DNA damage repair in iPSCs are responsible for their greater tendency towards mutagenesis. Methods: Mouse iPSCs, ESCs and embryonic fbroblasts were exposed to ionizing radiation (4 Gy) to introduce dou- ble-strand DNA breaks. At 4 h later, fdelity of DNA damage repair was assessed using whole-genome re-sequencing. We also analyzed genomic stability in mice derived from iPSCs versus ESCs. Results: In comparison to ESCs and embryonic fbroblasts, iPSCs had lower DNA damage repair capacity, more somatic mutations and short indels after irradiation. iPSCs showed greater non-homologous end joining DNA repair and less homologous recombination DNA repair. Mice derived from iPSCs had lower DNA damage repair capacity than ESC-derived mice as well as C57 control mice. Conclusions: The relatively low genomic stability of iPSCs and their high rate of tumorigenesis in vivo appear to be due, at least in part, to low fdelity of DNA damage repair. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

PIK-Sachbericht 2019
Inhaltsverzeichnis 01 Highlights 02 Eckdaten 03 Forschungsabteilungen 04 FutureLabs Wissenschaftsunterstützende 05 Organisationseinheiten 06 Anhang 7 United in Science 9 Von Deutschland nach Europa und in die Welt 12 Aus der Forschung 18 In eigener Sache 23 Wissenschaftliche Politikberatung 26 Medien-Highlights 2019 28 Besuche am PIK 29 Wissenschaftliche Politikberatung 30 Breitenwirkung 33 Klima, Kunst und Kultur 34 Berlin-Brandenburg – das PIK aktiv in der Heimat 36 Finanzierung | Beschäftigungszahlen 37 Publikationen | PIK in den Medien 38 Vorträge, Lehre und Veranstaltungen | Wissenschaftlicher Nachwuchs 40 Forschungsabteilung 1 – Erdsystemanalyse 46 Forschungsabteilung 2 – Klimaresilienz 52 Forschungsabteilung 3 – Transformationspfade 58 Forschungsabteilung 4 – Komplexitätsforschung 64 69 Informationstechnische Dienste 70 Verwaltung 71 Kommunikation 72 Stab der Direktoren 73 Wissenschaftsmanagement und Transfer 75 Organigramm 76 Kuratorium und Wissenschaftlicher Beirat 77 Auszeichnungen und Ernennungen 80 Berufungen, Habilitationen und Stipendien 81 Drittmittelprojekte 89 Veröff entlichungen 2019 5 Vorwort So klar man schon jetzt sagen kann, dass 2020 als Aber wir haben noch viel vor uns, das zeigt auch die das Corona-Jahr in die Geschichte eingehen wird, Pandemie-Krise, während derer dieser PIK-Sachbe- so klar lässt sich wohl auch sagen: 2019 war ein richt erstellt wurde. Die Herausforderungen werden Klima-Jahr. Klar wie nie zuvor standen Klimawandel komplexer und internationaler. Von den Planetaren und Klimapolitik im Mittelpunkt der öffentlichen Grenzen bis zu den Globalen Gemeinschaftsgütern: Aufmerksamkeit. Angestoßen durch die Fridays for Nachhaltiger Wohlstand im 21. Jahrhundert und da- Future-Bewegung gingen in Deutschland und überall rüber hinaus hängt ab vom grenzüberschreitenden auf der Welt Hunderttausende junge Menschen auf Management öff entlicher Güter – das gilt für den Ge- die Straße – unter Berufung auf die Klimaforschung, sundheitsschutz genauso wie für die Klimastabilität. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
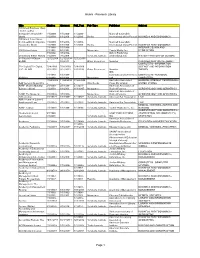
Research Library Page 1
Alumni - Research Library Title Citation Abstract Full_Text Pub Type Publisher Subject 100 Great Business Ideas : from Leading Companies Around the 1/1/2009- 1/1/2009- 1/1/2009- Marshall Cavendish World 1/1/2009 1/1/2009 1/1/2009 Books International (Asia) Pte Ltd BUSINESS AND ECONOMICS 100 Great Sales Ideas : from Leading Companies 1/1/2009- 1/1/2009- 1/1/2009- Marshall Cavendish Around the World 1/1/2009 1/1/2009 1/1/2009 Books International (Asia) Pte Ltd BUSINESS AND ECONOMICS 1/1/1988- 1/1/1988- INTERIOR DESIGN AND 1001 Home Ideas 6/1/1991 6/1/1991 Magazines Family Media, Inc. DECORATION 3/1/2002- 3/1/2002- Oxford Publishing 20 Century British History 7/1/2009 7/1/2009 Scholarly Journals Limited(England) HISTORY--HISTORY OF EUROPE 33 Charts [33 Charts - 12/12/2009 12/12/2009- 12/12/2009 BLOG] + 6/3/2011 + Other Resources Newstex CHILDREN AND YOUTH--ABOUT COMPUTERS--INFORMATION 50+ Digital [50+ Digital, 7/28/2009- 7/28/2009- 7/28/2009- SCIENCE AND INFORMATION LLC - BLOG] 2/22/2010 2/22/2010 2/22/2010 Other Resources Newstex THEORY IDG 1/1/1988- 1/1/1988- Communications/Peterboro COMPUTERS--PERSONAL 80 Micro 6/1/1988 6/1/1988 Magazines ugh COMPUTERS 11/24/2004 11/24/2004 11/24/2004 Australian Associated GENERAL INTEREST PERIODICALS-- AAP General News Wire + + + Wire Feeds Press Pty Limited UNITED STATES AARP Modern Maturity; 2/1/1988- 2/1/1988- 2/1/1991- American Association of [Library edition] 1/1/2003 1/1/2003 11/1/1997 Magazines Retired Persons GERONTOLOGY AND GERIATRICS American Association of AARP The Magazine 3/1/2003+ 3/1/2003+ Magazines Retired Persons GERONTOLOGY AND GERIATRICS ABA Journal 8/1/1972+ 1/1/1988+ 1/1/1992+ Scholarly Journals American Bar Association LAW ABA Journal of Labor & Employment Law 7/1/2007+ 7/1/2007+ 7/1/2007+ Scholarly Journals American Bar Association LAW MEDICAL SCIENCES--NURSES AND ABNF Journal 1/1/1999+ 1/1/1999+ 1/1/1999+ Scholarly Journals Tucker Publications, Inc. -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Stabilisation of hepatocyte phenotype using synthetic materials Baltasar Lucendo Villarin MRes University of Edinburgh 2015 This dissertation is submitted for the degree of Doctor of Philosophy Declaration This thesis is the result of my work and includes nothing that is the outcome of work done in collaboration, except where indicated in the text. The work in this thesis has not been submitted for any other degree or professional qualification. Baltasar Lucendo Villarin i ii Abstract Primary human hepatocytes are a scare resource with limited lifespan and variable function which diminishes with time in culture. As a consequence, their use in tissue modelling and therapy is restricted. Human embryonic stem cells (hESC) could provide a stable source of human tissue due to their self-renewal properties and their ability to give rise to all the cell types of the human body. -

I REGENERATIVE MEDICINE APPROACHES to SPINAL CORD
REGENERATIVE MEDICINE APPROACHES TO SPINAL CORD INJURY A Dissertation Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy Ashley Elizabeth Mohrman March 2017 i ABSTRACT Hundreds of thousands of people suffer from spinal cord injuries in the U.S.A. alone, with very few patients ever experiencing complete recovery. Complexity of the tissue and inflammatory response contribute to this lack of recovery, as the proper function of the central nervous system relies on its highly specific structural and spatial organization. The overall goal of this dissertation project is to study the central nervous system in the healthy and injured state so as to devise appropriate strategies to recover tissue homeostasis, and ultimately function, from an injured state. A specific spinal cord injury model, syringomyelia, was studied; this condition presents as a fluid filled cyst within the spinal cord. Molecular evaluation at three and six weeks post-injury revealed a large inflammatory response including leukocyte invasion, losses in neuronal transmission and signaling, and upregulation in important osmoregulators. These included osmotic stress regulating metabolites betaine and taurine, as well as the betaine/GABA transporter (BGT-1), potassium chloride transporter (KCC4), and water transporter aquaporin 1 (AQP1). To study cellular behavior in native tissue, adult neural stem cells from the subventricular niche were differentiated in vitro. These cells were tested under various culture conditions for cell phenotype preferences. A mostly pure (>80%) population of neural stem cells could be specified using soft, hydrogel substrates with a laminin coating and interferon-γ supplementation. -

Application of Fibrin-Based Hydrogels for Nerve Protection And
Yu et al. Journal of Biological Engineering (2020) 14:22 https://doi.org/10.1186/s13036-020-00244-3 REVIEW Open Access Application of fibrin-based hydrogels for nerve protection and regeneration after spinal cord injury Ziyuan Yu, Hongru Li, Peng Xia, Weijian Kong, Yuxin Chang, Chuan Fu, Kai Wang, Xiaoyu Yang* and Zhiping Qi* Abstract Traffic accidents, falls, and many other events may cause traumatic spinal cord injuries (SCIs), resulting in nerve cells and extracellular matrix loss in the spinal cord, along with blood loss, inflammation, oxidative stress (OS), and others. The continuous development of neural tissue engineering has attracted increasing attention on the application of fibrin hydrogels in repairing SCIs. Except for excellent biocompatibility, flexibility, and plasticity, fibrin, a component of extracellular matrix (ECM), can be equipped with cells, ECM protein, and various growth factors to promote damage repair. This review will focus on the advantages and disadvantages of fibrin hydrogels from different sources, as well as the various modifications for internal topographical guidance during the polymerization. From the perspective of further improvement of cell function before and after the delivery of stem cell, cytokine, and drug, this review will also evaluate the application of fibrin hydrogels as a carrier to the therapy of nerve repair and regeneration, to mirror the recent development tendency and challenge. Keywords: Fibrin hydrogels, Spinal cord injury, Topographical guidance, Stem cells delivery, Cytokines delivery, Drug delivery Introduction secondary death of remaining nerves or to enhance the The spinal cord is an integral part of the central nervous compensation of spared circuits function [4–7]. -

UC San Diego UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Insights from reconstructing cellular networks in transcription, stress, and cancer Permalink https://escholarship.org/uc/item/6s97497m Authors Ke, Eugene Yunghung Ke, Eugene Yunghung Publication Date 2012 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Insights from Reconstructing Cellular Networks in Transcription, Stress, and Cancer A dissertation submitted in the partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics and Systems Biology by Eugene Yunghung Ke Committee in charge: Professor Shankar Subramaniam, Chair Professor Inder Verma, Co-Chair Professor Web Cavenee Professor Alexander Hoffmann Professor Bing Ren 2012 The Dissertation of Eugene Yunghung Ke is approved, and it is acceptable in quality and form for the publication on microfilm and electronically ________________________________________________________________ ________________________________________________________________ ________________________________________________________________ ________________________________________________________________ Co-Chair ________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION To my parents, Victor and Tai-Lee Ke iv EPIGRAPH [T]here are known knowns; there are things we know we know. We also know there are known unknowns; that is to say we know there -

What Are IPS Cells? Stem Cell & Regenerative Medicine Center University of Wisconsin-Madison
What are IPS cells? Stem Cell & Regenerative Medicine Center University of Wisconsin-Madison n induced pluripotent stem cell, or IPS cell, How do we know induced pluripotent stem is a stem cell that has been created from an cells can match embryonic stem cells? So adult cell such as a skin, liver, stomach or far, induced pluripotent stem cells appear to Aother mature cell through the introduction of genes exhibit the same key features of embryonic that reprogram the cell and transform it into a cell stem cells: the ability to differentiate from a that has all the characteristics of an embryonic stem blank-slate state to any of the 220 types of cell. The term pluripotent connotes the ability of a cells in the human body, and the ability to cell to give rise to multiple cell types, including all reproduce indefinitely in culture. Because in- three embryonic lineages forming the body’s or- duced stem cells are relatively new, however, gans, nervous system, skin, muscle and skeleton. scientists must compare the cells to those obtained from embryos to assess their charac- What are the advantages of induced pluripotent teristics in detail and ensure that there are no stem cells? significant differences. Bioethics: Induced stem cells have the obvious edge Do induced pluripotent stem cells mean of not having to be derived from human embryos, we no longer need embryonic stem cells? a major ethical consideration. The ability to re- No. It remains to be seen whether repro- program an adult cell to behave like an embryonic grammed cells differ in significant ways from stem cell may also enable scientists to sidestep embryonic stem cells. -

Stem Cells: the Secret to Change | Science News for Kids
Stem cells: The secret to change | Science News for Kids http://www.sciencenewsforkids.org/2013/04/stem-cells-the-secret-to-change/ SNK E-Blast Sign-up Privacy Policy Contact Us AA BB OO UU TT SS NN KK CC OO MM PP EE TT EE Who We Are Broadcom MASTERS For Educators Our Sponsors Intel ISEF SSP News & Events Intel STS EXPLORE: ATOMS & FORCES EARTH & SKY HUMANS & HEALTH LIFE TECH & MATH EXTRA SEARCH FOR: HUMANS & HEALTH : BODY & HEALTH Stem cells: The secret to change Unusual, versatile cells hold the key to regrowing lost tissues By Alison Pearce Stevens / April 10, 2013 Related Links Deadly new virus emerges The AIDS virus that vanished Bad for breathing Sleeping in space Going Deeper P. B arry. “Stem cells, show your face.” Science News. August 24, 2008. Neurons created from induced stem cells in Iqbal Ahmad’s lab glow red with fluorescent dye. The neuroscientist at the University of Nebraska Medical Center is researching whether the nerve cells could one day help restore sight to patients S. Ornes. “The 2012 Nobel Prizes.” Science with glaucoma. Once injected in a patient, the nerve cells would work by inserting themselves between the retina and optic News for Kids. Oct. 19, 2012. nerve, restoring signals to the brain. Credit: Courtesy of Iqbal Ahmad Inside your body, red blood cells are constantly on the move. They deliver oxygen to every tissue in every E. Sohn. “From stem cell to any cell.” Science part of your body. These blood cells also cart away waste. So their work is crucial to your survival.