Physical Analysis of Human Hair
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

US5223252.Pdf
||||||||||||||| USOO522.3252A United States Patent (19) 11) Patent Number: 5,223,252 Kolc et al. 45 Date of Patent: Jun. 29, 1993 (54) PERMANENT WAVE COMPOSITION AND METHOD FOREIGN PATENT DOCUMENTS 344653 12/1989 European Pat. Off. 75) Inventors: Stanley J. Kolc, Chicago; Richard A. 352375 1/1990 European Pat. Off. Abbott, Westmont; Arun Nandagiri, Libertyville, all of Ill. OTHER PUBLICATIONS 73) Assignee: Helene Curtis, Inc., Chicago, Ill. JP 73 14934 B-english abstract. Primary Examiner-Thurman K. Page (21) Appl. No.: 919,972 Assistant Examiner-Neil Levy Attorney, Agent, or Firm-Marshall, O'Toole, Gerstein, 22 Filed: Jul. 27, 1992 Murray & Borun 57 ABSTRACT Related U.S. Application Data A mild, alkaline permanent wave reducing composition 63 Continuation of Ser. No. 670,056, Mar. 15, 1991, aban and method of permanently waving or reshaping doned. human hair that provides a strong, long lasting curl like an alkaline permanent wave composition but leaves the 51) Int. Cl. ................................................ A61K 7/09 hair soft like an acid permanent wave composition and 52 U.S. Cl. ........................................ 424/72; 424/71; leaves essentially no post-perm odor. The composition 131/203; 131/205 contains about 2.0% to about 6.5% by weight of a cys 58) Field of Search .................... 424/72, 71; 132/203, teine reducing agent compound selected from the group 132/204, 205, 209 consisting of cysteine, a cysteine salt, and mixtures (56) References Cited thereof; about 4.5% to about 8.0% of a thioglycolate; and sufficient additional alkali, if necessary, to bring the U.S. PATENT DOCUMENTS pH of the composition within the rang of about 7.5 to about 9.5. -

PRICELIST-1920-FINAL.Pdf
INDEX Page No. MD Speech 01 Our Vision / Our Mission 02 Product Classification and Grade Information 03 Label Information 04 GHS Compliance 05 Technical Data Sheet and COA 06 Qualikems Product Range 07 ISO Certificate 08 - 09 Company Details 10 Ordering Information 11 Terms & Conditions 12 Rate List 13 - 52 Images of Lab / Plant / R & D 53 - 58 Rate List 59 -116 BELIEVING yourselfIN IS THE FIRST SECRET TO Success Dear Reader, The document you are holding is the result of work performed by the team of professionals of QUALIKEMS. It is the fruit of our teams extensive technical experience combine with the collaboration of our customers, who have offered us their valuable comments and proposals for improvement. At Qualikems, we have been working and investing for many years with our thoughts focused on the long term. Only thus can this comprehensive catalogue be kept up to date with the products you need. Our highly trained workforce, using state of the art technology, is the driving force behind the management of our modern factory, and our principal aim is to guarantee that the QUALIKEMS product range meets the conditions you require. QUALIKEMS reinforces industrial character and the path to progress we have continuously forged over the years. This path requires the responsible use of resources and the sustainability of our business activity. It is likewise requires and ability to keep on growing as the way to earn and to preserve our status as the leading supplier of laboratory reagents to our Clients Ashok Sahni Managing Director QUALIKEMS FINE CHEM PVT. -

Gasket Chemical Services Guide
Gasket Chemical Services Guide Revision: GSG-100 6490 Rev.(AA) • The information contained herein is general in nature and recommendations are valid only for Victaulic compounds. • Gasket compatibility is dependent upon a number of factors. Suitability for a particular application must be determined by a competent individual familiar with system-specific conditions. • Victaulic offers no warranties, expressed or implied, of a product in any application. Contact your Victaulic sales representative to ensure the best gasket is selected for a particular service. Failure to follow these instructions could cause system failure, resulting in serious personal injury and property damage. Rating Code Key 1 Most Applications 2 Limited Applications 3 Restricted Applications (Nitrile) (EPDM) Grade E (Silicone) GRADE L GRADE T GRADE A GRADE V GRADE O GRADE M (Neoprene) GRADE M2 --- Insufficient Data (White Nitrile) GRADE CHP-2 (Epichlorohydrin) (Fluoroelastomer) (Fluoroelastomer) (Halogenated Butyl) (Hydrogenated Nitrile) Chemical GRADE ST / H Abietic Acid --- --- --- --- --- --- --- --- --- --- Acetaldehyde 2 3 3 3 3 --- --- 2 --- 3 Acetamide 1 1 1 1 2 --- --- 2 --- 3 Acetanilide 1 3 3 3 1 --- --- 2 --- 3 Acetic Acid, 30% 1 2 2 2 1 --- 2 1 2 3 Acetic Acid, 5% 1 2 2 2 1 --- 2 1 1 3 Acetic Acid, Glacial 1 3 3 3 3 --- 3 2 3 3 Acetic Acid, Hot, High Pressure 3 3 3 3 3 --- 3 3 3 3 Acetic Anhydride 2 3 3 3 2 --- 3 3 --- 3 Acetoacetic Acid 1 3 3 3 1 --- --- 2 --- 3 Acetone 1 3 3 3 3 --- 3 3 3 3 Acetone Cyanohydrin 1 3 3 3 1 --- --- 2 --- 3 Acetonitrile 1 3 3 3 1 --- --- --- --- 3 Acetophenetidine 3 2 2 2 3 --- --- --- --- 1 Acetophenone 1 3 3 3 3 --- 3 3 --- 3 Acetotoluidide 3 2 2 2 3 --- --- --- --- 1 Acetyl Acetone 1 3 3 3 3 --- 3 3 --- 3 The data and recommendations presented are based upon the best information available resulting from a combination of Victaulic's field experience, laboratory testing and recommendations supplied by prime producers of basic copolymer materials. -
Ammonia Free Perms
Ammonia free perms It seems the perm is back in the spotlight! More and more clients want hair that looks naturally curly. But one thing that's definitely NOT back? Shop from the world's largest selection and best deals for Ammonia-Free Hair Perms. Shop with confidence on eBay! More than 40% highlighted. SOFT curl. FIRM curl. Extra Care. Ammonia-Free/Low Thio. PERMS. Classic. Body. Acid Perm. Extra. Body. Firm Acid Perm. Texture. You know the saying “you always want what you can't have?” Well, that is exactly me when it comes to curly hair. At age 11, I was putting my. This technique uses ThinkCurl™ from Organic Colour Systems, which uses different chemistry than a traditional. One and Only Low Ammonia Texturizing Acid Perm leaves hair with superior shine and silky softness. FREE SHIPPING ON ANY $50 PURCHASE. Here is what you need to know about "thio-free" perms and how they compare to traditional perms. How does perming work? Your hair is made. Yes you can find Ammonia-Free Perming System services! Choosing ammonia free perming products is getting easier than ever before. Organic Curl Systems is. Ammonia-Free waves use an ingredient that does not evaporate as readily as ammonia, Thio-Free Waves use a substitute for Ammonium Thioglycolate (ATG) as the primary reducing agent in the waving formula. Non-smelling perm. Ammonia free perm. Imagine that your innately straight hair becomes curly, a bit like an angel's tresses. Show me a woman with straight hair that did not. However, you should keep in mind that acid perms, and low-ammonia/ammonia-free perms are going to give you curls that are less “firm” than their alkaline. -

Flowchem VE Corrosion Protection 2014
General Overview Flowchem VE Corrosion Resistance Guide Chemical Environment Recommended Temperature Limit °C WATER 100 ACIDS INORGANIC 100 ORGANIC 100 OXIDISING 65 ALKALINE INORGANIC 70 ORGANIC 50 SALTS INORGANIC 100 ORGANIC 50 SOLVENTS ALKYL, >C6 80 AROMATIC 25 ALCOHOLS 65 AMINES 25 HALOGENS 25 COPY KETONES NR NOTES: 1. General overview, exceptions within each group are possible. However increasing the molecular weight of chemicals will generally increase the service temperature. 2. This information is just a global grouping. Service temperature within one family can vary between different chemicals. 3. All temperatures are given for aqueous solutions. In case of concentrated mixtures service temperatures are generally lower. 4. Discoloration/ staining is not classified as chemical attack if hardness is unchanged. 5. Higher temperatures will reduce the chemical resistance shown in the performance table. 6. Some chemicals may concentrate due to evaporation and become more aggressive. 7. Mixtures of chemicals can be more aggressive than might be expected from the individual components alone. Note: The data contained herein is based on laboratory tests performed under carefully controlled conditions. No warranty can be expressed or implied regarding the accuracy of this information, as it will apply to actual operational use. Plant operations vary widely, and the individual results obtained are affected by the specific conditions encountered, which are beyond our control. Important: UNCONTROLLEDWe believe the information contained here to be true and accurate as of the date of publication. Flowcrete makes no warranty, expressed or implied, based on this literature and assumes no responsibility for consequential or incidental damages in the use of the systems described, including any warranty of merchantability or fitness. -

This Generic Chemical Compatibility Chart Is Offered Only As a General
Chemical Compatibility Chart This Generic Chemical Compatibility Chart is offered only as a general guide, indicating the suitability of various elastomers for service in these chemicals and fluids. The ratings are based, for the most part, on published literature of various polymer suppliers, rubber manufacturers, and in some cases, the considered opinion of experienced compounders. We cannot guarantee their accuracy nor assume any responsibility for use thereof. Therefore, we recommend that you always TEST your materials for your particular application before moving to production. www.GlobalORing.com 4250 N. Sam Houston Parkway E, Suite 100, Houston, TX 77032 Phone: 832-448-5550 / 877-448-5550 Fax: 832-448-5551 / 877-448-5551 Chemical Compatibility Chart Polymer Comparison Temperature Durometer Media Resistance ASTM Range (°F) Range Abbr Low High Low High Oil Fuel Acid Alkali Ozone Steam Brake Weather ACM -40 400 50 80 E P – G P -F G - E G - E P P E AEM -50 350 40 90 F P – G P – F P – F E P P E AU -65 250 40 90 G P – G P – G P – F E P P E CIIR -50 300 60 70 P P F – E G - E E G – E G F CM -60 300 80 80 G – E P – F G – E E E P - F G E CR -80 220 30 90 F – G P – F F – G G – P G – E P – G F P - G CSM -65 275 50 80 E F G – E G – E E – O P – G F E – O ECO -80 to -15 275 to 325 50 90 E G - E P - G P – G G – E F – G P G – E EPDM -75 350 40 90 P P G G G - E E G – E E EU -65 250 40 90 G P – G P – G P – F E P P E FEPM 0 450 to 500 70 90 E P - F F - G E E G - E F – E E FKM -20 to 20 500 50 90 E E G - E P - G O P – G F E FVMQ -110 to -90 450 40 -

Office 2,990,336
Office 2,990,336 Paterated June 27, 196 2 step of employing the hair waving components com 2,990,336 prising water soluble mercaptains in carbon dioxide gen PROCESS OF HAR WAVING ANB) erating solutions, such as carbonic acid water whereby Harry Martin,PREPARATION Monroe, N.Y., THEREFOR assignor, by direct and introduction of harmful minerals in the hair waving mesne assignments to Permanent Hair Waving Corpo 5 preparation is minimized and the introduction of oxidiz ration, Norfolk, Va., a corporation of V ing components retarded, if not completely eliminated. No Drawing. Filed Oct. 16, 1959, Ser. No. 846,818 Still more particularly it is contemplated by my in 9 Clains. (C.167-87.) vention to provide a hair waving preparation which is This invention relates to compounds and methods for self contained in the relationship of the mercaptain radical hair waving and preparations therefor. 10 to the base, which may be stored under any climatic con Known to me are the methods for waving hair in the ditions without deterioration so as not to require special cold by the use of mercapto products and more particu packaging prescriptions but which is arranged to be larly such products as thioglycerine, thioglycolic acid and brought into solution in an aqueous medium when re a wide range of mercaptain containing materials disclosed quired by the addition of the aqueous solvent, whereby in my prior applications Serial No. 383,204, now Patent 15 the formation of the undesirable by-products and ob No. 2,350,178, and Serial No. 437,628, filed April 4, 1942, noxious odors is minimized, if not completely avoided, now Patent No. -

EPDM & FKM Chemical Resistance Guide
EPDM & FKM Chemical Resistance Guide FIRST EDITION EPDM & FKM CHEMICAL RESISTANCE GUIDE Elastomers: Ethylene Propylene (EPDM) Fluorocarbon (FKM) Chemical Resistance Guide Ethylene Propylene (EPDM) & Fluorocarbon (FKM) 1st Edition © 2019 by IPEX. All rights reserved. No part of this book may be used or reproduced in any manner whatsoever without prior written permission. For information contact: IPEX, Marketing, 1425 North Service Road East, Oakville, Ontario, Canada, L6H 1A7 ABOUT IPEX At IPEX, we have been manufacturing non-metallic pipe and fittings since 1951. We formulate our own compounds and maintain strict quality control during production. Our products are made available for customers thanks to a network of regional stocking locations from coast-to-coast. We offer a wide variety of systems including complete lines of piping, fittings, valves and custom-fabricated items. More importantly, we are committed to meeting our customers’ needs. As a leader in the plastic piping industry, IPEX continually develops new products, modernizes manufacturing facilities and acquires innovative process technology. In addition, our staff take pride in their work, making available to customers their extensive thermoplastic knowledge and field experience. IPEX personnel are committed to improving the safety, reliability and performance of thermoplastic materials. We are involved in several standards committees and are members of and/or comply with the organizations listed on this page. For specific details about any IPEX product, contact our customer service department. INTRODUCTION Elastomers have outstanding resistance to a wide range of chemical reagents. Selecting the correct elastomer for an application will depend on the chemical resistance, temperature and mechanical properties needed. Resistance is a function both of temperatures and concentration, and there are many reagents which can be handled for limited temperature ranges and concentrations. -
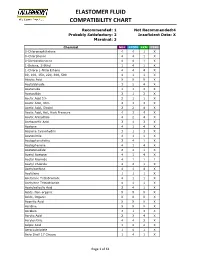
Elastomer Fluid Compatibility Chart
ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE 0-Chloronaphthalene 4 4 1 X 0-Chlorphenol 4 4 1 X 0-Dichlorobenzene 4 4 1 X 1-Butene, 2-Ethyl 1 4 1 X 1-Chloro 1-Nitro Ethane 4 4 4 X 90, 100, 150, 220, 300, 500 4 1 1 X Abietic Acid X X X X Acetaldehyde 3 2 4 X Acetamide 1 1 3 X Acetanilide 3 1 3 X Acetic Acid 5% 2 1 1 X Acetic Acid, 30% 2 1 3 X Acetic Acid, Glacial 2 2 4 X Acetic Acid, Hot, High Pressure 4 3 4 X Acetic Anhydride 4 2 4 X Acetoacetic Acid 3 1 3 X Acetone 4 1 4 X Acetone Cyanohydrin 3 1 3 X Acetonitrile 3 1 1 X Acetophenetidine 2 4 1 X Acetophenone 4 1 4 X Acetotoluidide 2 4 1 X Acetyl Acetone 4 1 4 X Acetyl Bromide 4 1 1 1 Acetyl Chloride 4 4 1 X Acetylacetone 4 1 4 X Acetylene 1 1 1 X Acetylene Tetrabromide 4 1 1 X Acetylene Tetrachloride 4 1 1 X Acetylsalicylic Acid 2 4 1 X Acids, Non-organic X X X X Acids, Organic X X X X Aconitic Acid X X X X Acridine X X X X Acrolein 3 1 3 X Acrylic Acid 2 3 4 X Acrylonitrile 4 4 3 X Adipic Acid 1 2 2 X Aero Lubriplate 1 4 1 X Aero Shell 17 Grease 1 4 1 X Page 1 of 61 ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE Aero Shell 1AC Grease 1 4 1 X Aero Shell 750 2 4 1 X Aero Shell 7A Grease 1 4 1 X Aerosafe 2300 4 1 4 X Aerosafe 2300W 4 1 4 X Aerozene 50 (50% Hydrazine 50% UDMH) 3 1 4 X Air, 1 1 1 X Air, 200 - 300° F 3 2 1 X Air, 300 - 400° F 4 4 1 X Air, 400 - 500° F 4 4 3 X Air, -

2 Answer All the Questions. 1 Hydrogen, H , Reacts
CHERRY HILL TUITION OCR A CHEMISTRY A2 PAPER 26 2 Answer all the questions. 1 Hydrogen, H2, reacts with nitrogen monoxide, NO, as shown in the equation below. 2H2(g) + 2NO(g) N2(g) + 2H2O(g) A chemist carries out a series of experiments and determines the rate equation for this reaction: 2 rate = k[H2(g)][NO(g)] (a) In one of the experiments, the chemist reacts together: –2 –3 • 1.2 × 10 mol dm H2(g) • 6.0 × 10–3 mol dm–3 NO(g) The initial rate of this reaction is 3.6 × 10–2 mol dm–3 s –1. Calculate the rate constant, k, for this reaction. State the units, if any. k = ……………………… units ……………………… [3] (b) Predict what would happen to the initial rate of reaction for the following changes in concentrations. (i) The concentration of H2(g) is doubled. ...................................................................................................................................... [1] (ii) The concentration of NO(g) is halved. ...................................................................................................................................... [1] (iii) The concentrations of H2(g) and NO(g) are both increased by four times. ........................................................................................................................................... ...................................................................................................................................... [1] © OCR 2011 1 CHERRY HILL TUITION OCR A CHEMISTRY A2 PAPER 26 3 (c) The chemist carries out the reaction between hydrogen and nitrogen -

Product List 2013-14 Cat
RANGE OF LABORATORY CHEMICALS PRODUCT LIST 2013-14 CAT. NOS. PRODUCT NAME CAS.NOS. PACKING A-00141 ABSCISIC ACID 99% (For Biochemistry) 21293-29-8 25 mg (ABA) 100 mg A-00151 ACACIA (Confirming to IP) 9000-01-05 500 gm (Gum Acacia Powder ) 10 x 500 gm A-00158 ACENAPHTHENE 96% (For Synthesis) 83-32-9 100 gm 500 gm A-00160 ACES BUFFER 99% 7365-82-4 25 gm [N-2-Acetamido-2- Aminoethane Sulphonic Acid] 100 gm A-00163 ACETALDEHYDE SOLUTION 20-30% 75-07-0 500 ml 2.5 Ltr. A-00168 ACETAMIDE 99% (For Synthesis) 60-35-5 500 gm A-00170 ACETAMIDINIUM HYDROCHLORIDE 124-42-5 100 gm (Acetamidium Chloride) A-00178 ACETANILIDE 98.5 % (For Synthesis) 103-84-4 500 gm (N-Phenyl Acetamide) 10 x 500 gm A-00180 ACETATE BUFFER SOLUTION PH 4.6 126-96-5 500 ml A-00188 ACETIC ACID GLACIAL (For Synthesis) 64-19-7 500 ml 2.5Ltr. ACETIC ACID GLACIAL 99.7% AR 64-19-7 500 ml A-00188/100 2.5 lit. A-00189 ACETIC ACID METHYLAMIDE 79-16-3 100 ml A-00190 ACETIC ACID SOLUTION 0.1N --------- 500 ml A-00191 ACETIC ACID SOLUTION 1%, 2%, 3% --------- 500ml A-00193 ACETO ACETANILIDE 98% (For Synthesis) 102-01-2 500 gm A-00199 O-ACETO ACETANISIDIDE 98% 92-15-9 100 gm A-00205 ACETO CARMINE SOLUTION --------- 100 ml (For Microscopical Staining) 500 ml A-00206 ACETO CARMINE SOLUTION AR --------- 100 ml 500 ml A-00210 ACETONE 99% (EX - PURE) 67-64-1 500 ml (For Synthesis) 25 x 500 ml 2.5Ltr. -

Click Here (PDF)
Chemical Products Caustic Soda, Liquid (Gluconated) Fumaric Acid Monoaluminum Phosphate Cetyl Alcohol Monoammonium Phosphate Acetic Acid Chromic Acid Monocalcium Phosphate Acetic Anhydride CIP Cleaner Monoethanolamine (MEA) Acetone Citric Acid (Dry or Liquid) Gluconate Liquid Monopotassium Phosphate Activated Carbon Cocamide DEA Gluconic Acid Monosodium Glutamate (MSG) Alcohol Ethoxylates (91-6, 91-8, etc) Cocamide MEA Glycerine (Crude, Tech, or USP) Monosodium Phosphate Alkanolamides Cocamidopropyl Betaine Glycol Ether, “E” Series Muriate of Potash (KCL) Alkyl Ether Sulfates Copper Cyanide Glycol Ether, “P” Series Muriatic Acid (HCL) Alkyl Sulfates Copper Nitrate Glycol Stearate Aloe Vera Copper Sulfate Glycolic Acid Alpha Methyl Styrene (AMS) Cosmetic Oils Aluminum Chloride Cyclohexane Nickel Chloride Aluminum Stearate Cyclohexanone Nickel Sulfate Aluminum Sulfate (Dry or Liquid) Cyclohexylamine Heptane Nitric Acid Amido-Amines Hexane N-Methyl Pyrrolidone Amine Oxides Hexylene Glycol NPE Surfactant (NP-4, NP-6, NP-9) Ammonia, Aqua Hydrazine N-Propyl Acetate Ammonium Bifluoride Dequest® Hydrochloric Acid N-Propyl Alcohol Ammonium Chloride Diacetone Alcohol (Inhibited Available) NTA Ammonium Lauryl Sulfate Diammonium Phosphate Hydrofluoric Acid Ammonium Persulfate Diatomaceous Earth Hydrofluosilicic Acid Ammonium Sulfate Dibasic Esters Hydrogen Peroxide Ammonium Thioglycolate Dicalcium Phosphate Hydroxyacetic Acid Oxalic Acid Ammonium Thiosulfate Dicalite® Hydroxylamine Sulfate Amphoterics Diethanolamine (DEA) Hypophosphorous Acid Amyl