Approval Letter
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

FDA Comments on CBD in Foods
Popular Content Public Health Focus FDA and Marijuana: Questions and Answers 1. How is marijuana therapy being used by some members of the medical community? 2. Why hasn’t the FDA approved marijuana for medical uses? 3. Is marijuana safe for medical use? 4. How does FDA’s role differ from NIH and DEA’s role when it comes to the investigation of marijuana for medical use? 5. Does the FDA object to the clinical investigation of marijuana for medical use? 6. What kind of research is the FDA reviewing when it comes to the efficacy of marijuana? 7. How can patients get into expanded access program for marijuana for medical use? 8. Does the FDA have concerns about administering a cannabis product to children? 9. What is FDA’s reaction to states that are allowing marijuana to be sold for medical uses without the FDA’s approval? 10. What is the FDA’s position on state “Right to Try” bills? 11. Has the agency received any adverse event reports associated with marijuana for medical conditions? 12. Can products that contain cannabidiol be sold as dietary supplements? 13. Is it legal, in interstate commerce, to sell a food to which cannabidiol has been added? 14. In making the two previous determinations, why did FDA conclude that substantial clinical investigations have been authorized for and/or instituted about cannabidiol, and that the existence of such investigations has been made public? 15. Will FDA take enforcement action regarding cannabidiol products that are marketed as dietary supplements? What about foods to which cannabidiol has been added? 16. -
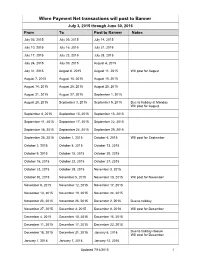
Transactions Posted to Pathway
When Payment Net transactions will post to Banner July 3, 2015 through June 30, 2016 From To Post to Banner Notes July 03, 2015 July 09, 2015 July 14, 2015 July 10, 2015 July 16, 2015 July 21, 2015 July 17, 2015 July 23, 2015 July 28, 2015 July 24, 2015 July 30, 2015 August 4, 2015 July 31, 2015 August 6, 2015 August 11, 2015 Will post for August August 7, 2015 August 13, 2015 August 18, 2015 August 14, 2015 August 20, 2015 August 25, 2015 August 21, 2015 August 27, 2015 September 1, 2015 August 28, 2015 September 3, 2015 September 9, 2015 Due to holiday at Monday Will post for August September 4, 2015 September 10, 2015 September 15, 2015 September 11, 2015 September 17, 2015 September 22, 2015 September 18, 2015 September 24, 2015 September 29, 2015 September 25, 2015 October 1, 2015 October 6, 2015 Will post for September October 2, 2015 October 8, 2015 October 13, 2015 October 9, 2015 October 15, 2015 October 20, 2015 October 16, 2015 October 22, 2015 October 27, 2015 October 23, 2015 October 29, 2015 November 3, 2015 October 30, 2015 November 5, 2015 November 10, 2015 Will post for November November 6, 2015 November 12, 2015 November 17, 2015 November 13, 2015 November 19, 2015 November 24, 2015 November 20, 2015 November 26, 2015 December 2, 2015 Due to holiday November 27, 2015 December 3, 2015 December 8, 2018 Will post for December December 4, 2015 December 10, 2015 December 15, 2015 December 11, 2015 December 17, 2015 December 22, 2015 December 18, 2015 December 31, 2015 January 6, 2016 Due to holiday closure Will post for December -
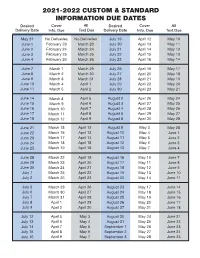
2021-2022 Custom & Standard Information Due Dates
2021-2022 CUSTOM & STANDARD INFORMATION DUE DATES Desired Cover All Desired Cover All Delivery Date Info. Due Text Due Delivery Date Info. Due Text Due May 31 No Deliveries No Deliveries July 19 April 12 May 10 June 1 February 23 March 23 July 20 April 13 May 11 June 2 February 24 March 24 July 21 April 14 May 12 June 3 February 25 March 25 July 22 April 15 May 13 June 4 February 26 March 26 July 23 April 16 May 14 June 7 March 1 March 29 July 26 April 19 May 17 June 8 March 2 March 30 July 27 April 20 May 18 June 9 March 3 March 31 July 28 April 21 May 19 June 10 March 4 April 1 July 29 April 22 May 20 June 11 March 5 April 2 July 30 April 23 May 21 June 14 March 8 April 5 August 2 April 26 May 24 June 15 March 9 April 6 August 3 April 27 May 25 June 16 March 10 April 7 August 4 April 28 May 26 June 17 March 11 April 8 August 5 April 29 May 27 June 18 March 12 April 9 August 6 April 30 May 28 June 21 March 15 April 12 August 9 May 3 May 28 June 22 March 16 April 13 August 10 May 4 June 1 June 23 March 17 April 14 August 11 May 5 June 2 June 24 March 18 April 15 August 12 May 6 June 3 June 25 March 19 April 16 August 13 May 7 June 4 June 28 March 22 April 19 August 16 May 10 June 7 June 29 March 23 April 20 August 17 May 11 June 8 June 30 March 24 April 21 August 18 May 12 June 9 July 1 March 25 April 22 August 19 May 13 June 10 July 2 March 26 April 23 August 20 May 14 June 11 July 5 March 29 April 26 August 23 May 17 June 14 July 6 March 30 April 27 August 24 May 18 June 15 July 7 March 31 April 28 August 25 May 19 June 16 July 8 April 1 April 29 August 26 May 20 June 17 July 9 April 2 April 30 August 27 May 21 June 18 July 12 April 5 May 3 August 30 May 24 June 21 July 13 April 6 May 4 August 31 May 25 June 22 July 14 April 7 May 5 September 1 May 26 June 23 July 15 April 8 May 6 September 2 May 27 June 24 July 16 April 9 May 7 September 3 May 28 June 25. -

Vaccines, Diagnostics, and Treatments): Frequently Asked Questions
Development and Regulation of Medical Countermeasures for COVID-19 (Vaccines, Diagnostics, and Treatments): Frequently Asked Questions June 25, 2020 Congressional Research Service https://crsreports.congress.gov R46427 SUMMARY R46427 Development and Regulation of Medical June 25, 2020 Countermeasures for COVID-19 (Vaccines, Agata Dabrowska Diagnostics, and Treatments): Frequently Analyst in Health Policy Asked Questions Frank Gottron Specialist in Science and In recent months, the Coronavirus Disease 2019 (COVID-19) pandemic has spread globally, with Technology Policy the United States now reporting the highest number of cases of any country in the world. Currently, there are few treatment options available to lessen the health impact of the disease and no vaccines or other prophylactic treatments to curb the spread of the virus. Amanda K. Sarata Specialist in Health Policy The biomedical community has been working to develop new therapies or vaccines, and to repurpose already approved therapeutics, that could prevent COVID-19 infections or lessen Kavya Sekar severe outcomes in patients. In addition, efforts have been underway to develop new diagnostic Analyst in Health Policy tools (i.e., testing) to help better identify and isolate positive cases, thereby reducing the spread of the disease. To this end, Congress has appropriated funds for research and development into new medical countermeasures (MCMs) in several recent supplemental appropriations acts. MCMs are medical products that may be used to treat, prevent, or diagnose conditions associated with emerging infectious diseases or chemical, biological, radiological, or nuclear (CBRN) agents. MCMs include biologics (e.g., vaccines, monoclonal antibodies), drugs (e.g., antimicrobials, antivirals), and medical devices (e.g., diagnostic tests). -

Comparisons of Food and Drug Administration and European
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector VALUE IN HEALTH 15 (2012) 1108–1118 Available online at www.sciencedirect.com journal homepage: www.elsevier.com/locate/jval Health Policy Analyses Comparisons of Food and Drug Administration and European Medicines Agency Risk Management Implementation for Recent Pharmaceutical Approvals: Report of the International Society for Pharmacoeconomics and Outcomes Research Risk Benefit Management Working Group Yvonne Lis, PhD1, Melissa H. Roberts, MS2, Shital Kamble, PhD3, Jeff J. Guo, PhD4, Dennis W. Raisch, PhD5,* 1PAREXEL International, Uxbridge, UK; 2Lovelace Clinic Foundation Research, Albuquerque, New Mexico, USA; 3Quintiles-Outcome, Rockville, MD, USA; 4College of Pharmacy, University of Cincinnati Medical Center, Cincinnati, OH, USA; 5College of Pharmacy, University of New Mexico, Albuquerque, NM, USA ABSTRACT Objective: 1) To compare the Food and Drug Administration’s (FDA’s) requirements not included in RMPs were patient medication guides Risk Evaluation and Mitigation Strategies (REMS) and European (100% of the drugs), provider communication plans (38%), and routine Medicines Agency’s (EMA’s) Risk Management Plan (RMP) guidances monitoring of REMS (66%). RMP requirements not included in REMS and 2) to compare REMS and RMPs for specific chemical entities and were specific adverse event reporting (45% of the drugs), prospective biological products. Methods: FDA, EMA, and pharmaceutical com- registry studies (34%), prospective epidemiology studies (24%), addi- pany Web sites were consulted for details pertaining to REMS and tional trial data (28%), and Summary of Product Characteristics RMPs. REMS requirements include medication guides, communication contraindications (76%). Conclusions: Both REMS and RMPs provide plans, elements to ensure safe use, implementation systems, and positive guidance for identification, monitoring, and minimization of specified assessment intervals. -

New Drugs Are Not Enough‑Drug Repositioning in Oncology: an Update
INTERNATIONAL JOURNAL OF ONCOLOGY 56: 651-684, 2020 New drugs are not enough‑drug repositioning in oncology: An update ROMINA GABRIELA ARMANDO, DIEGO LUIS MENGUAL GÓMEZ and DANIEL EDUARDO GOMEZ Laboratory of Molecular Oncology, Science and Technology Department, National University of Quilmes, Bernal B1876, Argentina Received August 15, 2019; Accepted December 16, 2019 DOI: 10.3892/ijo.2020.4966 Abstract. Drug repositioning refers to the concept of discov- 17. Lithium ering novel clinical benefits of drugs that are already known 18. Metformin for use treating other diseases. The advantages of this are that 19. Niclosamide several important drug characteristics are already established 20. Nitroxoline (including efficacy, pharmacokinetics, pharmacodynamics and 21. Nonsteroidal anti‑inflammatory drugs toxicity), making the process of research for a putative drug 22. Phosphodiesterase-5 inhibitors quicker and less costly. Drug repositioning in oncology has 23. Pimozide received extensive focus. The present review summarizes the 24. Propranolol most prominent examples of drug repositioning for the treat- 25. Riluzole ment of cancer, taking into consideration their primary use, 26. Statins proposed anticancer mechanisms and current development 27. Thalidomide status. 28. Valproic acid 29. Verapamil 30. Zidovudine Contents 31. Concluding remarks 1. Introduction 2. Artesunate 1. Introduction 3. Auranofin 4. Benzimidazole derivatives In previous decades, a considerable amount of work has been 5. Chloroquine conducted in search of novel oncological therapies; however, 6. Chlorpromazine cancer remains one of the leading causes of death globally. 7. Clomipramine The creation of novel drugs requires large volumes of capital, 8. Desmopressin alongside extensive experimentation and testing, comprising 9. Digoxin the pioneer identification of identifiable targets and corrobora- 10. -

COVID-19 Dashboard - Friday, August 21, 2020 Dashboard of Public Health Indicators
8/21/2020 Duplicate of Public Health Indicators Massachusetts Department of Public Health COVID-19 Dashboard - Friday, August 21, 2020 Dashboard of Public Health Indicators Newly Reported Total Confirmed Newly Reported Total Deaths Confirmed Cases Cases Deaths among among Confirmed Today Confirmed Today Cases 431 115,741 13 8,670 New Individuals Total Individuals Below is the current status: Tested by Tested by Molecular Tests Molecular Tests Measure Status 26,758 1,535,862 COVID-19 positive test rate ⚫ Number of individuals who died from COVID-19 ⚫ Total Molecular Legend Number of patients with COVID-19 in hospitals ⚫ Tests Healthcare system readiness ⚫ Administered Testing capacity ⚫ 2,050,871 Contact tracing capabilities ⚫ The front page of the dashboard has been reformatted. Antibody tests (individual and total numbers) can be found on page 17. 1 1/1 8/21/2020 Public Health Indicators2 Massachusetts Department of Public Health COVID-19 Dashboard - Friday, August 21, 2020 Percent or Count of Change Since Dashboard of Public Health Indicators Lowest Observed Value (LOV) 7 Day Weighted 1.7% 1.6% 1.6% 1.5% 1.5% 1.7% 1.7% 1.8% 1.8% 1.7% 1.7% 1.7% 1.7% 1.4% 1.4% 1.4% Average of Positive 1.5% 1.7% 1.7% 1.7% 1.6% 1.6% 1.6% 1.6% 1.4% 1.6% 1.6% 1.3% 1.2% 0% Molecular Test Rate* 1.4% 1.0% 1.2% 22 23 24 25 26 27 28 29 30 31 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 July August LOV =1.2% 3 Day Average of 500 Number of 427 465 396 396 403 407 COVID-19 Patients 383 384 393 384 391 382 0% 400 373 377 368374 368 374 378 375 380 381 371 371 -

2021 7 Day Working Days Calendar
2021 7 Day Working Days Calendar The Working Day Calendar is used to compute the estimated completion date of a contract. To use the calendar, find the start date of the contract, add the working days to the number of the calendar date (a number from 1 to 1000), and subtract 1, find that calculated number in the calendar and that will be the completion date of the contract Date Number of the Calendar Date Friday, January 1, 2021 133 Saturday, January 2, 2021 134 Sunday, January 3, 2021 135 Monday, January 4, 2021 136 Tuesday, January 5, 2021 137 Wednesday, January 6, 2021 138 Thursday, January 7, 2021 139 Friday, January 8, 2021 140 Saturday, January 9, 2021 141 Sunday, January 10, 2021 142 Monday, January 11, 2021 143 Tuesday, January 12, 2021 144 Wednesday, January 13, 2021 145 Thursday, January 14, 2021 146 Friday, January 15, 2021 147 Saturday, January 16, 2021 148 Sunday, January 17, 2021 149 Monday, January 18, 2021 150 Tuesday, January 19, 2021 151 Wednesday, January 20, 2021 152 Thursday, January 21, 2021 153 Friday, January 22, 2021 154 Saturday, January 23, 2021 155 Sunday, January 24, 2021 156 Monday, January 25, 2021 157 Tuesday, January 26, 2021 158 Wednesday, January 27, 2021 159 Thursday, January 28, 2021 160 Friday, January 29, 2021 161 Saturday, January 30, 2021 162 Sunday, January 31, 2021 163 Monday, February 1, 2021 164 Tuesday, February 2, 2021 165 Wednesday, February 3, 2021 166 Thursday, February 4, 2021 167 Date Number of the Calendar Date Friday, February 5, 2021 168 Saturday, February 6, 2021 169 Sunday, February -
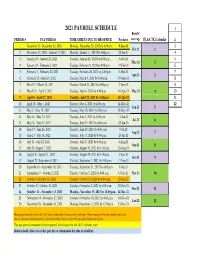
Payroll Calendar 2021
2021 PAYROLL SCHEDULE 1 Benefit PERIOD # PAY PERIOD TIME SHEETS DUE TO HR OFFICE Paydates coverage FLSA 7K Calendar 2 1 December 13- December 26, 2020 Monday, December 28, 2020 by 4:00 p.m. 8-Jan-21 3 Feb-21 1 2 December 27, 2020 - Janurary 9, 2021 Monday, January 11, 2021 by 4:00 p.m. 22-Jan-21 4 3 January 10 - January 23, 2021 Tuesday, January 26, 2021 by 4:00 p.m. 5-Feb-21 5 Mar-21 2 4 January 24 - February 6, 2021 Tuesday, February 9, 2021 by 4:00 p.m. 19-Feb-21 6 5 February 7 - February 20, 2021 Tuesday, February 26, 2021 by 4:00 p.m. 5-Mar-21 7 Apr-21 3 6 February 21 - March 6, 2021 Tuesday, March 9, 2021 by 4:00 p.m. 19-Mar-21 8 7 March 7 - March 20, 2021 Tuesday, March 23, 2021 by 4:00 p.m. 2-Apr-21 9 8 March 21 - April 3, 2021 Tuesday, April 6, 2021 by 4:00 p.m. 16-Apr-21 May-21 4 10 9 April 4 - April 17, 2021 Tuesday, April 20, 2021 by 4:00 p.m. 30-Apr-21 11 10 April 18 - May 1, 2021 Tuesday, May 4, 2021 by 4:00 p.m. 14-May-21 12 Jun-21 5 11 May 2 - May 15, 2021 Tuesday, May 18, 2021 by 4:00 p.m. 28-May-21 12 May 16 - May 29, 2021 Tuesday, June 1, 2021 by 4:00 p.m. 11-Jun-21 Jul-21 6 13 May 30 - June 12, 2021 Tuesday, June 15, 2021 by 4:00 p.m. -

Drug Development and Clinical Trials 101
DRUG DEVELOPMENT AND CLINICAL TRIALS 101 March 27, 2021 A Resource of PWSA | USA | pwsausa.org DISCOVERY - PHASE 1 Dean S. Carson, PhD, Saniona, Inc. A Resource of PWSA | USA | pwsausa.org DRUG DISCOVERY AND DEVELOPMENT Why is it important? • Rare hormone producing tumor of the anterior pituitary gland • Cabergoline, an ergot derivative, is a potent dopamine D2 receptor agonist that blocks pituitary production of prolactin • Ergot is a fungus that grows on rye and related plants, and produces alkaloids that can cause ergotism in humans and animals that eat the contaminated grain • Painful seizures, mania, psychosis, headaches, nausea, vomiting A Resource of PWSA | USA | pwsausa.org 3 PHARMACOLOGY • Pharmacology is the science of how • Clinical Pharmacology applies the basic drugs and biologics act on biological science and principles of pharmacology to systems and how the system responds to the practice of medicine the drug Basic science Translational pharmacology Clinical practice Doctors, pharmacists, dentists, veterinarians use the information discovered from pharmacology to select the best medications to treat patients A Resource of PWSA | USA | pwsausa.org 4 MECHANISM OF ACTION • Modern drug discovery is a mechanism • What are the known consequences of that driven field mechanism in a disease state? ie. • Nearly all new experimental therapeutics potential efficacy have a defined mechanism-of-action and • What are the known consequences of that most have companion diagnostics allowing mechanism in healthy systems? ie. researchers to -

Research and Development in the Pharmaceutical Industry
Research and Development in the Pharmaceutical Industry APRIL | 2021 At a Glance This report examines research and development (R&D) by the pharmaceutical industry. Spending on R&D and Its Results. Spending on R&D and the introduction of new drugs have both increased in the past two decades. • In 2019, the pharmaceutical industry spent $83 billion dollars on R&D. Adjusted for inflation, that amount is about 10 times what the industry spent per year in the 1980s. • Between 2010 and 2019, the number of new drugs approved for sale increased by 60 percent compared with the previous decade, with a peak of 59 new drugs approved in 2018. Factors Influencing R&D Spending. The amount of money that drug companies devote to R&D is determined by the amount of revenue they expect to earn from a new drug, the expected cost of developing that drug, and policies that influence the supply of and demand for drugs. • The expected lifetime global revenues of a new drug depends on the prices that companies expect to charge for the drug in different markets around the world, the volume of sales they anticipate at those prices, and the likelihood the drug-development effort will succeed. • The expected cost to develop a new drug—including capital costs and expenditures on drugs that fail to reach the market—has been estimated to range from less than $1 billion to more than $2 billion. • The federal government influences the amount of private spending on R&D through programs (such as Medicare) that increase the demand for prescription drugs, through policies (such as spending for basic research and regulations on what must be demonstrated in clinical trials) that affect the supply of new drugs, and through policies (such as recommendations for vaccines) that affect both supply and demand. -

2021-2022 JECA School Calendar
July 2021 2021-2022 January 2022 S M T W T F S JECA School Calendar S M T W T F S 1 2 3 1 4 5 6 7 8 9 10 2 3 4 [5 6 7 8 11 12 13 14 15 16 17 9 10 11 12 13 14 15 18 19 20 21 22 23 24 16 17 18 19 20 21 22 Professional Development 25 26 27 28 29 30 31 August 2-4 November 12 23 24 25 26 27 28 29 September 24 January 3 30 31 August 2021 October 8 March 11 October 29 (NLC) February 2022 S M T W T F S Teacher Work Days 1 2 3 4 5 6 7 S M T W T F S August 5-10 January 4 1 2 3 4 5 8 9 10 [11 12 13 14 December 17 May 20-24 15 16 17 18 19 20 21 6 7 8 9 10 11 12 Staff and Student Holidays 13 14 15 16 17 18 19 22 23 24 25 26 27 28 Labor Day Sept 6 20 21 22 23 24 25 26 29 30 31 Thanksgiving Nov 25-26 Winter Break Dec 20 - 31 27 28 MLK Jan 17 September 2021 Spring Break Mar 14-18 March 2022 S M T W T F S Battle of Flowers Apr 8 S M T W T F S 1 2 3 4 Good Friday Apr 15 1 2 3 4 5 5 6 7 8 9 10 11 Important Dates 6 7 8 9 10] 11 12 12 13 14 15 16 17 18 JECA classes begin August 10 Seniors begin S1* August 16 13 14 15 16 17 18 19 19 20 21 22 23 24 25 NLC classes begin August 23 20 [21 22 23 24 25 26 26 27 28 29 30 NLC S1 Exams December 6-10 27 28 29 30 31 Seniors end S1* December 10 October 2021 JECA Semester Exams December 13-16 nd April 2022 S M T W T F S JECA 2 semester begins January 5 Seniors begin S2* January 10 S M T W T F S 1 2 NLC classes begin January 18 1 2 3 4 5 6 7] 8 9 NLC S2 Exams May 9-13 10 [11 12 13 14 15 16 Last day for Seniors* May 13 3 4 5 6 7 8 9 17 18 19 20 21 22 23 JECA Final Exams May 16-19 10 11 12 13 14 15 16 Last day of school May