Androgen Receptor-Dependent Mechanisms Mediating Drug Resistance in Prostate Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CHAPTER 29 ORGANIC CHEMICALS VI 29-1 Notes 1
)&f1y3X CHAPTER 29 ORGANIC CHEMICALS VI 29-1 Notes 1. Except where the context otherwise requires, the headings of this chapter apply only to: (a) Separate chemically defined organic compounds, whether or not containing impurities; (b) Mixtures of two or more isomers of the same organic compound (whether or not containing impurities), except mixtures of acyclic hydrocarbon isomers (other than stereoisomers), whether or not saturated (chapter 27); (c) The products of headings 2936 to 2939 or the sugar ethers and sugar esters, and their salts, of heading 2940, or the products of heading 2941, whether or not chemically defined; (d) Products mentioned in (a), (b) or (c) above dissolved in water; (e) Products mentioned in (a), (b) or (c) above dissolved in other solvents provided that the solution constitutes a normal and necessary method of putting up these products adopted solely for reasons of safety or for transport and that the solvent does not render the product particularly suitable for specific use rather than for general use; (f) The products mentioned in (a), (b), (c), (d) or (e) above with an added stabilizer (including an anticaking agent) necessary for their preservation or transport; (g) The products mentioned in (a), (b), (c), (d), (e) or (f) above with an added antidusting agent or a coloring or odoriferous substance added to facilitate their identification or for safety reasons, provided that the additions do not render the product particularly suitable for specific use rather than for general use; (h) The following products, diluted to standard strengths, for the production of azo dyes: diazonium salts, couplers used for these salts and diazotizable amines and their salts. -

Expression of Aromatase, Estrogen Receptor and , Androgen
0031-3998/06/6006-0740 PEDIATRIC RESEARCH Vol. 60, No. 6, 2006 Copyright © 2006 International Pediatric Research Foundation, Inc. Printed in U.S.A. Expression of Aromatase, Estrogen Receptor ␣ and , Androgen Receptor, and Cytochrome P-450scc in the Human Early Prepubertal Testis ESPERANZA B. BERENSZTEIN, MARI´A SONIA BAQUEDANO, CANDELA R. GONZALEZ, NORA I. SARACO, JORGE RODRIGUEZ, ROBERTO PONZIO, MARCO A. RIVAROLA, AND ALICIA BELGOROSKY Research Laboratory [E.B.B., M.S.B., C.R.G., N.I.S., J.R., M.A.R., A.B.], Hospital de Pediatria Garrahan, Buenos Aires C124 5AAM, Argentina; Centro de Investigaciones en Reproduccion [R.P.], Facultad de Medicina, Universidad de Buenos Aires, Buenos Aires C112 1ABG, Argentina ABSTRACT: The expression of aromatase, estrogen receptor ␣ might affect adult testicular cell mass, as well as testicular (ER␣) and  (ER), androgen receptor (AR), and cytochrome P-450 function (8). side chain cleavage enzyme (cP450scc) was studied in prepubertal In humans (9), there are three growth phases of LCs testis. Samples were divided in three age groups (GRs): GR1, during testicular development. Fetal LCs produce testoster- ϭ newborns (1- to 21-d-old neonates, n 5); GR2, postnatal activation one required for fetal masculinization and Insl-3, necessary ϭ stage (1- to 7-mo-old infants, n 6); GR3, childhood (12- to for testicular descent (10). They regress during the third ϭ ␣ 60-mo-old boys, n 4). Absent or very poor detection of ER by trimester of pregnancy. A second wave of infantile LCs has immunohistochemistry in all cells and by mRNA expression was been described during the postnatal surge of luteinizing observed. -

Ricini Oleum
PHARMACOGNOSY II PHAR306 6th Semester 5th Lecture Prof. Dr. Müberra Koşar Ass. Prof. Dr. Aybike Yektaoğlu Eastern Mediterranean University Faculty of Pharmacy Department of Pharmacognosy PHARMACEUTICAL FIXED OILS AND ANIMAL FATS FIXED OILS & ANIMAL FATS Amygdalae oleum • “Almond oil” • obtained by crushing of the seeds of two varieties Prunus dulcis var. dulcis or P. dulcis var. amara (Rosaceae) in the cold • Almond oil is obtained in the Mediterranean countries (Italy, France, Spain and North Africa) where its culture is obtained • The only difference between the two varieties is the cyanogenic glycoside content of the var. amara FIXED OILS&ANIMAL FATS Amygdalae oleum • seeds carries 40-55% fixed oil • the refined oil mainly contains oleic acid (62-86%), linoleic (20- 30%), palmitic (4-9%) • Amydalae oleum raffinatum (Almond oil, refined) (Eur.Pu.) • Amydalae oleum virginale (Almond oil, virgin) (Eur.Ph.) • major used in cosmetology and dermatology • used as a carrier in oily injectable preparations FIXED OILS&ANIMAL FATS Arachidis oleum • “Arachis oil, Peanut oil” – “Peanut butter” • Arachis hypogaea (Fabaceae) • cultivated in South America, China, India, Australia, and West Africa • due to various genotypes they vary in fatty acid content • the seeds are cold-pressed • they have similar properties as olive oil • most suitable oil for added for embedding purposes into other oils (e.g. olive oil) FIXED OILS&ANIMAL FATS Arachidis oleum - content • seeds carries 40-50% fixed oil • 50-65% oleic acid • 18-30% linoleic acid • 8-10% palmitic -

Influence of Androgen Receptor on the Prognosis of Breast Cancer
Journal of Clinical Medicine Article Influence of Androgen Receptor on the Prognosis of Breast Cancer 1, , 2, 1 2 3 Ki-Tae Hwang * y , Young A Kim y , Jongjin Kim , Jeong Hwan Park , In Sil Choi , Kyu Ri Hwang 4 , Young Jun Chai 1 and Jin Hyun Park 3 1 Department of Surgery, Seoul Metropolitan Government Seoul National University Boramae Medical Center, 39, Boramae-Gil, Dongjak-gu, Seoul 156-707, Korea; [email protected] (J.K.); [email protected] (Y.J.C.) 2 Department of Pathology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul 156-707, Korea; [email protected] (Y.A.K.); [email protected] (J.H.P.) 3 Department of Internal Medicine, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul 156-707, Korea; [email protected] (I.S.C.); [email protected] (J.H.P.) 4 Department of Obstetrics & Gynecology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul 156-707, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-2-870-2275; Fax: +82-2-831-2826 These authors contributed equally to this work. y Received: 28 February 2020; Accepted: 8 April 2020; Published: 10 April 2020 Abstract: We investigated the prognostic influence of androgen receptor (AR) on breast cancer. AR status was assessed using immunohistochemistry with tissue microarrays from 395 operable primary breast cancer patients who received curative surgery. The Kaplan–Meier estimator was used to analyze the survival rates and a log-rank test was used to determine the significance of the differences in survival. The Cox proportional hazards model was used to calculate the hazard ratio (HR) and the 95% confidence interval (CI) of survival. -

Immunohistochemical Study of Androgen, Estrogen and Progesterone Receptors in Salivary Gland Tumors
Oral Pathology Oral Pathology Immunohistochemical study of androgen, estrogen and progesterone receptors in salivary gland tumors Fabio Augusto Ito(a) Abstract: The aim of this work was to study the immunohistochemi- (b) Kazuhiro Ito cal expression of androgen receptor, estrogen receptor and progesterone Ricardo Della Coletta(c) Pablo Agustín Vargas(c) receptor in pleomorphic adenomas, Warthin’s tumors, mucoepidermoid Márcio Ajudarte Lopes(c) carcinomas and adenoid cystic carcinomas of salivary glands. A total of 41 pleomorphic adenomas, 30 Warthin’s tumors, 30 mucoepidermoid carcinomas and 30 adenoid cystic carcinomas were analyzed, and the im- (a) DDS, PhD; (b)MD, Professor – Department of Pathology, Londrina State University, munohistochemical expression of these hormone receptors were assessed. Londrina, PR, Brazil. It was observed that all cases were negative for estrogen and progesterone (c) DDS, PhD, Professor, Department of Oral receptors. Androgen receptor was positive in 2 cases each of pleomorphic Diagnosis, Piracicaba Dental School, University of Campinas (UNICAMP), adenoma, mucoepidermoid carcinoma and adenoid cystic carcinoma. In Piracicaba, SP, Brazil. conclusion, the results do not support a role of estrogen and progesterone in the tumorigenesis of pleomorphic adenomas, Warthin’s tumors, muco- epidermoid carcinomas and adenoid cystic carcinomas. However, andro- gen receptors can play a role in a small set of salivary gland tumors, and this would deserve further studies. Descriptors: Receptors, androgen; Receptors, estrogen; Receptors, progesterone; Salivary gland neoplasms. Corresponding author: Márcio Ajudarte Lopes Semiologia, Faculdade de Odontologia de Piracicaba, UNICAMP Av. Limeira, 901 Caixa Postal: 52 CEP: 13414-903 Piracicaba - SP - Brazil E-mail: [email protected] Received for publication on Oct 01, 2008 Accepted for publication on Sep 22, 2009 Braz Oral Res. -

The Natural Compounds Atraric Acid and N-Butylbenzene-Sulfonamide
The natural compounds atraric acid and N-butylbenzene-sulfonamide as antagonists of the human androgen receptor and inhibitors of prostate cancer cell growth Daniela Roell, Aria Baniahmad To cite this version: Daniela Roell, Aria Baniahmad. The natural compounds atraric acid and N-butylbenzene-sulfonamide as antagonists of the human androgen receptor and inhibitors of prostate cancer cell growth. Molec- ular and Cellular Endocrinology, Elsevier, 2010, 332 (1-2), pp.1. 10.1016/j.mce.2010.09.013. hal- 00654968 HAL Id: hal-00654968 https://hal.archives-ouvertes.fr/hal-00654968 Submitted on 24 Dec 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Accepted Manuscript Title: The natural compounds atraric acid and N-butylbenzene-sulfonamide as antagonists of the human androgen receptor and inhibitors of prostate cancer cell growth Authors: Daniela Roell, Aria Baniahmad PII: S0303-7207(10)00474-0 DOI: doi:10.1016/j.mce.2010.09.013 Reference: MCE 7644 To appear in: Molecular and Cellular Endocrinology Received date: 29-6-2010 Revised date: 3-9-2010 Accepted date: 27-9-2010 Please cite this article as: Roell, D., Baniahmad, A., The natural compounds atraric acid and N-butylbenzene-sulfonamide as antagonists of the human androgen receptor and inhibitors of prostate cancer cell growth, Molecular and Cellular Endocrinology (2010), doi:10.1016/j.mce.2010.09.013 This is a PDF file of an unedited manuscript that has been accepted for publication. -

Androgen Receptor Is Targeted to Distinct Subcellular Compartments in Response to Different Therapeutic Antiandrogens
7392 Vol. 10, 7392–7401, November 1, 2004 Clinical Cancer Research Androgen Receptor Is Targeted to Distinct Subcellular Compartments in Response to Different Therapeutic Antiandrogens Hayley C. Whitaker,1 Sarah Hanrahan,3 informed sequential treatment regime may benefit prostate Nick Totty,3 Simon C. Gamble,1 cancer patients. The observed subnuclear and subcytoplas- Jonathan Waxman,1 Andrew C. B. Cato,4 mic associations of the AR suggest new areas of study to 2 1 investigate the role of the AR in the response and resistance Helen C. Hurst, and Charlotte L. Bevan of prostate cancer to antiandrogen therapy. 1Prostate Cancer Research Group and 2Cancer Research UK Molecular Oncology Unit, Department of Cancer Medicine, Faculty of Medicine, Imperial College London, London, United Kingdom; INTRODUCTION 3Protein Analysis, Cancer Research UK, London Research Institute, Prostate cancer is the most commonly diagnosed male London, United Kingdom; and 4Forschungszentrum Karlsruhe, cancer in the United States and the second leading cause of male Institute of Toxicology and Genetics, Karlsruhe, Germany cancer death (1). Prostate growth is initially androgen depend- ent; thus, treatment involves reducing circulating androgen lev- ABSTRACT els using leuteinizing hormone-releasing hormone analogs and opposing androgen action using antiandrogens. Little is known Purpose: Antiandrogens are routinely used in the treat- about how antiandrogens elicit their effects, although they act at ment of prostate cancer. Although they are known to pre- the level of the androgen receptor (AR), which mediates the vent activation of the androgen receptor (AR), little is effects of all androgens including the major circulating androgen known about the mechanisms involved. This report repre- testosterone and the more potent dihydrotestosterone (DHT). -

The Role of the Androgen Receptor Signaling in Breast Malignancies PANAGIOTIS F
ANTICANCER RESEARCH 37 : 6533-6540 (2017) doi:10.21873/anticanres.12109 Review The Role of the Androgen Receptor Signaling in Breast Malignancies PANAGIOTIS F. CHRISTOPOULOS*, NIKOLAOS I. VLACHOGIANNIS*, CHRISTIANA T. VOGKOU and MICHAEL KOUTSILIERIS Department of Experimental Physiology, School of Medicine, National and Kapodistrian University of Athens, Athens, Greece Abstract. Breast cancer (BrCa) is the most common decades, BrCa still has a poor prognosis with 5-year survival malignancy among women worldwide, and one of the leading rates of metastatic disease reaching to 26% only. BrCa is the causes of cancer-related deaths in females. Despite the second leading cause of death among female cancers with development of novel therapeutic modalities, triple-negative 40,610 estimated deaths in the U.S. expected in 2017 (1). breast cancer (TNBC) remains an incurable disease. Androgen Breast cancer comprises a heterogeneous group of diseases receptor (AR) is widely expressed in BrCa and its role in the with variable course and outcome. Currently, BrCa is sub- disease may differ depending on the molecular subtype and the classified into distinct molecular subtypes named: normal stage. Interestingly, AR has been suggested as a potential target breast like, luminal A/B, HER-2 related, basal-like and claudin- candidate in TNBC, while sex hormone levels may regulate the low (2, 3). Estrogen receptor (ER), progesterone receptor (PR) role of AR in BrCa subtypes. In the presence of estrogen and HER2 have long been established as useful prognostic and receptor α ( ERa ), AR may antagonize the ER α- induced effects, predictive biomarkers. Hormonal therapy in ER and PR whereas in the absence of estrogens, AR may act as an ER α- positive tumors (4), as well as the use of monoclonal antibodies mimic, promoting tumor. -

Annex 2B Tariff Schedule of the United States See General Notes to Annex 2B for Staging Explanation HTSUS No
Annex 2B Tariff Schedule of the United States See General Notes to Annex 2B for Staging Explanation HTSUS No. Description Base Rate Staging 0101 Live horses, asses, mules and hinnies: 0101.10.00 -Purebred breeding animals Free E 0101.90 -Other: 0101.90.10 --Horses Free E 0101.90.20 --Asses 6.8% B --Mules and hinnies: 0101.90.30 ---Imported for immediate slaughter Free E 0101.90.40 ---Other 4.5% A 0102 Live bovine animals: 0102.10.00 -Purebred breeding animals Free E 0102.90 -Other: 0102.90.20 --Cows imported specially for dairy purposes Free E 0102.90.40 --Other 1 cent/kg A 0103 Live swine: 0103.10.00 -Purebred breeding animals Free E -Other: 0103.91.00 --Weighing less than 50 kg each Free E 0103.92.00 --Weighing 50 kg or more each Free E 0104 Live sheep and goats: 0104.10.00 -Sheep Free E 0104.20.00 -Goats 68 cents/head A 0105 Live poultry of the following kinds: Chickens, ducks, geese, turkeys and guineas: -Weighing not more than 185 g: 0105.11.00 --Chickens 0.9 cents each A 0105.12.00 --Turkeys 0.9 cents each A 0105.19.00 --Other 0.9 cents each A -Other: 0105.92.00 --Chickens, weighing not more than 2,000 g 2 cents/kg A 0105.93.00 --Chickens, weighing more than 2,000 g 2 cents/kg A 0105.99.00 --Other 2 cents/kg A 0106 Other live animals: -Mammals: 0106.11.00 --Primates Free E 0106.12.00 --Whales, dolphins and porpoises (mammals of the order Cetacea); manatees and dugongs (mammals of the order Sirenia) Free E 0106.19 --Other: 2B-Schedule-1 HTSUS No. -

Estrogen Receptor Β, a Regulator of Androgen Receptor Signaling in The
Estrogen receptor β, a regulator of androgen receptor PNAS PLUS signaling in the mouse ventral prostate Wan-fu Wua, Laure Maneixa, Jose Insunzab, Ivan Nalvarteb, Per Antonsonb, Juha Kereb, Nancy Yiu-Lin Yub, Virpi Tohonenb, Shintaro Katayamab, Elisabet Einarsdottirb, Kaarel Krjutskovb, Yu-bing Daia, Bo Huanga, Wen Sua,c, Margaret Warnera, and Jan-Åke Gustafssona,b,1 aCenter for Nuclear Receptors and Cell Signaling, University of Houston, Houston, TX 77204; bCenter for Innovative Medicine, Department of Biosciences and Nutrition, Karolinska Institutet, Novum, 14186 Stockholm, Sweden; and cAstraZeneca-Shenzhen University Joint Institute of Nephrology, Centre for Nephrology & Urology, Shenzhen University Health Science Center, Shenzhen 518060, China Contributed by Jan-Åke Gustafsson, March 31, 2017 (sent for review February 8, 2017; reviewed by Gustavo E. Ayala and David R. Rowley) − − − − As estrogen receptor β / (ERβ / ) mice age, the ventral prostate (13). Several ERβ-selective agonists have been synthesized (14– (VP) develops increased numbers of hyperplastic, fibroplastic le- 20), and they have been found to be antiinflammatory in the brain sions and inflammatory cells. To identify genes involved in these and the gastrointestinal tract (21, 22) and antiproliferative in cell changes, we used RNA sequencing and immunohistochemistry to lines (23–27) and cancer models (23, 28). We have previously − − compare gene expression profiles in the VP of young (2-mo-old) shown that there is an increase in p63-positive cells in ERβ / − − and aging (18-mo-old) ERβ / mice and their WT littermates. We mouse VP but that these cells were not confined to the basal layer also treated young and old WT mice with an ERβ-selective agonist but were interdispersed with the basal and luminal layer (13). -
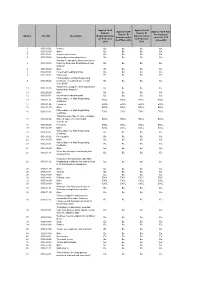
Number HS 2007 Description Applied Tariff Rate for Guatemala Under The
Applied Tariff Applied Tariff Applied Tariff Applied Tariff Rate Rate for Rate for El Rate for El for Honduras Number HS 2007 Description Guatemala under Salvador under Salvador under under the FTA the FTA since the FTA since the FTA in 2010 since 2010 2010 2011 1 0101.10.01 Horses Ex. Ex. Ex. Ex. 2 0101.10.99 Other Ex. Ex. Ex. Ex. 3 0101.90.01 Jump or race horses Ex. Ex. Ex. Ex. 4 0101.90.02 Non pedigreed breeding horses Ex. Ex. Ex. Ex. "Horses for slaughter, when imported by 5 0101.90.03 Federally Inspected Establishment type Ex. Ex. Ex. Ex. packers." 6 0101.90.99 Other Ex. Ex. Ex. Ex. 7 0102.10.01 Pure-bred breeding animals Ex. Ex. Ex. Ex. 8 0102.90.01 Dairy cows Ex. Ex. Ex. Ex. "With pedigree or High Registrating 9 0102.90.02 Certificate, excluding those of code Ex. Ex. Ex. Ex. 0102.90.01" "Bovines for slaughter, when imported by 10 0102.90.03 Ex. Ex. Ex. Ex. Industrial de Abastos." 11 0102.90.99 Other Ex. Ex. Ex. Ex. 12 0103.10.01 Pure-bred breeding animals Ex. Ex. Ex. Ex. With pedigree or High Registrating 13 0103.91.01 EXCL. EXCL. EXCL. EXCL. Certificate. 14 0103.91.02 Peccaries EXCL. EXCL. EXCL. EXCL. 15 0103.91.99 Other EXCL. EXCL. EXCL. EXCL. With pedigree or High Registrating 16 0103.92.01 EXCL. EXCL. EXCL. EXCL. Certificate. "Weighing more than 110 kg., excluding 17 0103.92.02 those of codes 0103.92.01 and EXCL. -

• Lipids: a Heterogeneous Class of Naturally Occurring Organic
Organic Lecture Series 1 Organic Lecture Series •• Lipids:Lipids: a heterogeneous class of naturally occurring organic compounds classified together on the basis of common solubility properties –they are insoluble in water but soluble in aprotic organic solvents, including diethyl ether, methylene chloride, and acetone 2 Organic Lecture Series • Lipids include –triglycerides, phospholipids, prostaglandins, prostacyclins, and fat-soluble vitamins –cholesterol, steroid hormones, and bile acids 3 Organic Lecture Series TriglyceridesTriglycerides •• Triglyceride:Triglyceride: an ester of glycerol with three fatty acids O CH OCR O 2 CH 2 OH RCOOH 1. NaOH, H2 O R' COCH O HOCH + R'COOH 2 . HCl, H O 2 CH OH R' ' COOH CH 2 OCR'' 2 A triglyceride 1,2,3-Propanetriol Fatty acids (Glycerol, glycerin) 4 FattyFatty acidacid Organic Lecture Series • Fatty acid: a carboxylic acid derived from hydrolysis of animal fats, vegetable oils, or membrane phospholipids – nearly all have an even number of carbon atoms, most between 12 and 20, in an unbranched chain – the three most abundant are palmitic (16:0), stearic acid (18:0), and oleic acid (18:1) 5 FattyFatty acidacid Organic Lecture Series –the three most abundant are palmitic (16:0), stearic acid (18:0), and oleic acid (18:1) –In this labeling system: (carbon #:alkene #) palmitate (16:0) oleate (18:1) O O CH 2 OC(CH2 ) 14CH3 stearate (18:0) CH3 (CH2 )7 CH= CH( CH2 )7 COCH O CH 2 OC(CH2 ) 16CH3 6 FattyFatty acidacid Organic Lecture Series –in most unsaturated fatty acids, the cis isomer predominates; the