Batten Disease: Biochemical and Molecular Characterization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Palmitoyl-Protein Thioesterase 1 Deficiency in Drosophila Melanogaster Causes Accumulation
Genetics: Published Articles Ahead of Print, published on February 1, 2006 as 10.1534/genetics.105.053306 Palmitoyl-protein thioesterase 1 deficiency in Drosophila melanogaster causes accumulation of abnormal storage material and reduced lifespan Anthony J. Hickey*,†,1, Heather L. Chotkowski*, Navjot Singh*, Jeffrey G. Ault*, Christopher A. Korey‡,2, Marcy E. MacDonald‡, and Robert L. Glaser*,†,3 * Wadsworth Center, New York State Department of Health, Albany, NY 12201-2002 † Department of Biomedical Sciences, State University of New York, Albany, NY 12201-0509 ‡ Molecular Neurogenetics Unit, Center for Human Genetic Research, Massachusetts General Hospital, Boston, MA 02114 1 current address: Albany Medical College, Albany, NY 12208 2 current address: Department of Biology, College of Charleston, Charleston, SC 294243 3 corresponding author: Wadsworth Center, NYS Dept. Health, P. O. Box 22002, Albany, NY 12201-2002 E-mail: [email protected] 1 running title: Phenotypes of Ppt1-deficient Drosophila key words: Batten disease infantile neuronal ceroid lipofuscinosis palmitoyl-protein thioesterase CLN1 Drosophila corresponding author: Robert L. Glaser Wadsworth Center, NYS Dept. Health P. O. Box 22002 Albany, NY 12201-2002 E-mail: [email protected] phone: 518-473-4201 fax: 518-474-3181 2 ABSTRACT Human neuronal ceroid lipofuscinoses (NCLs) are a group of genetic neurodegenerative diseases characterized by progressive death of neurons in the central nervous system (CNS) and accumulation of abnormal lysosomal storage material. Infantile NCL (INCL), the most severe form of NCL, is caused by mutations in the Ppt1 gene, which encodes the lysosomal enzyme palmitoyl-protein thioesterase 1 (Ppt1). We generated mutations in the Ppt1 ortholog of Drosophila melanogaster in order to characterize phenotypes caused by Ppt1-deficiency in flies. -

Diagnosis of Neuronal Ceroid Lipofuscinosis Type 2 (CLN2 Disease): Expert Recommendations for Early Detection and Laboratory Diagnosis
Molecular Genetics and Metabolism 119 (2016) 160–167 Contents lists available at ScienceDirect Molecular Genetics and Metabolism journal homepage: www.elsevier.com/locate/ymgme Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis Michael Fietz a, Moeenaldeen AlSayed b, Derek Burke c, Jessica Cohen-Pfeffer d,JonathanD.Coopere, Lenka Dvořáková f, Roberto Giugliani g, Emanuela Izzo d, Helena Jahnová f,ZoltanLukacsh,SaraE.Molei, Ines Noher de Halac j,DavidA.Pearcek,HelenaPoupetovaf, Angela Schulz l, Nicola Specchio m, Winnie Xin n, Nicole Miller d,⁎ a Department of Diagnostic Genomics, PathWest Laboratory Medicine WA, Nedlands, Australia b Department of Medical Genetics, Alfaisal University, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia c Chemical Pathology, Camelia Botnar Laboratories, Great Ormond Street Hospital, London, UK d BioMarin Pharmaceutical Inc., Novato, CA, USA e Institute of Psychiatry, Psychology & Neuroscience, King's College London, London, UK f Institute of Inherited Metabolic Disorders, First Faculty of Medicine, Charles University in Prague, General University Hospital in Prague, Prague, Czech Republic g Medical Genetics Service, HCPA, Department of Genetics, UFRGS, INAGEMP, Porto Alegre, Brazil h Newborn Screening and Metabolic Diagnostics Unit, Hamburg University Medical Center, Hamburg, Germany i MRC Laboratory for Molecular Cell Biology, UCL Institute of Child Health, University College London, London, UK j -

The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z
REVIEW pubs.acs.org/CR The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z. Long* and Benjamin F. Cravatt* The Skaggs Institute for Chemical Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States CONTENTS 2.4. Other Phospholipases 6034 1. Introduction 6023 2.4.1. LIPG (Endothelial Lipase) 6034 2. Small-Molecule Hydrolases 6023 2.4.2. PLA1A (Phosphatidylserine-Specific 2.1. Intracellular Neutral Lipases 6023 PLA1) 6035 2.1.1. LIPE (Hormone-Sensitive Lipase) 6024 2.4.3. LIPH and LIPI (Phosphatidic Acid-Specific 2.1.2. PNPLA2 (Adipose Triglyceride Lipase) 6024 PLA1R and β) 6035 2.1.3. MGLL (Monoacylglycerol Lipase) 6025 2.4.4. PLB1 (Phospholipase B) 6035 2.1.4. DAGLA and DAGLB (Diacylglycerol Lipase 2.4.5. DDHD1 and DDHD2 (DDHD Domain R and β) 6026 Containing 1 and 2) 6035 2.1.5. CES3 (Carboxylesterase 3) 6026 2.4.6. ABHD4 (Alpha/Beta Hydrolase Domain 2.1.6. AADACL1 (Arylacetamide Deacetylase-like 1) 6026 Containing 4) 6036 2.1.7. ABHD6 (Alpha/Beta Hydrolase Domain 2.5. Small-Molecule Amidases 6036 Containing 6) 6027 2.5.1. FAAH and FAAH2 (Fatty Acid Amide 2.1.8. ABHD12 (Alpha/Beta Hydrolase Domain Hydrolase and FAAH2) 6036 Containing 12) 6027 2.5.2. AFMID (Arylformamidase) 6037 2.2. Extracellular Neutral Lipases 6027 2.6. Acyl-CoA Hydrolases 6037 2.2.1. PNLIP (Pancreatic Lipase) 6028 2.6.1. FASN (Fatty Acid Synthase) 6037 2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic 2.6.2. -

Human Palmitoyl-Protein Thioesterase 1 (PPT1) ELISA Kit
Product Datasheet Human Palmitoyl-protein thioesterase 1 (PPT1) ELISA Kit Catalog No: #EK8355 Orders: [email protected] Package Size: #EK8355-1 48T #EK8355-2 96T Support: [email protected] Description Product Name Human Palmitoyl-protein thioesterase 1 (PPT1) ELISA Kit Brief Description ELISA Kit Applications ELISA Species Reactivity Human (Homo sapiens) Other Names RP11-115D7.2; CLN1; INCL; PPT; ceroid-palmitoyl-palmitoyl-protein thioesterase 1|palmitoyl-protein hydrolase 1 Accession No. P50897 Storage The stability of ELISA kit is determined by the loss rate of activity. The loss rate of this kit is less than 5% within the expiration date under appropriate storage condition. The loss rate was determined by accelerated thermal degradation test. Keep the kit at 37C for 4 and 7 days, and compare O.D.values of the kit kept at 37C with that of at recommended temperature. (referring from China Biological Products Standard, which was calculated by the Arrhenius equation. For ELISA kit, 4 days storage at 37C can be considered as 6 months at 2 - 8C, which means 7 days at 37C equaling 12 months at 2 - 8C). Application Details Detect Range:1.56-100 pg/mL Sensitivity:0.72 pg/mL Sample Type:Serum, Plasma, Other biological fluids Sample Volume: 1-200 µL Assay Time:1-4.5h Detection wavelength:450 nm Product Description Detection Method:SandwichTest principle:This assay employs a two-site sandwich ELISA to quantitate PPT1 in samples. An antibody specific for PPT1 has been pre-coated onto a microplate. Standards and samples are pipetted into the wells and anyPPT1 present is bound by the immobilized antibody. -

Molecular Interactions Underlying Neuronal Ceroid Lipofuscinoses CLN1 and CLN5
Annina Lyly Molecular Interactions Underlying Neuronal Ceroid Lipofuscinoses CLN1 and CLN5 Publications of the National Public Health Institute A 17/2008 Department of Molecular Medicine National Public Health Institute and Faculty of Medicine, University of Helsinki, Finland Helsinki, Finland 2008 Annina Lyly MOLECULAR INTERACTIONS UNDERLYING NEURONAL CEROID LIPOFUSCINOSES CLN1 AND CLN5 ACADEMIC DISSERTATION To be presented with the permission of the Medical Faculty of the University of Helsinki, for public examination in the Niilo Hallman lecture hall, Hospital for Children and Adolescents, Helsinki University Central Hospital, on June 6th , 2008, at 12 noon. National Public Health Institute, Helsinki, Finland and Faculty of Medicine, University of Helsinki, Finland Helsinki 2008 Publications of the National Public Health Institute KTL A17 / 2008 Copyright National Public Health Institute Julkaisija-Utgivare-Publisher Kansanterveyslaitos (KTL) Mannerheimintie 166 00300 Helsinki Puh. vaihde (09) 474 41, telefax (09) 4744 8408 Folkhälsoinstitutet Mannerheimvägen 166 00300 Helsingfors Tel. växel (09) 474 41, telefax (09) 4744 8408 National Public Health Institute Mannerheimintie 166 FIN-00300 Helsinki, Finland Telephone +358 9 474 41, telefax +358 9 4744 8408 ISBN 978-951-740-821-9 ISSN 0359-3584 ISBN 978-951-740-822-6 (pdf) ISSN 1458-6290 (pdf) Kannen kuva - cover graphic: Annina Lyly Yliopistopaino Helsinki 2008 Supervised by Adjunct Professor Anu Jalanko Department of Molecular Medicine National Public Health Institute, and Institute for Molecular Medicine Finland Helsinki, Finland Adjunct Professor Aija Kyttälä Department of Molecular Medicine National Public Health Institute, and Institute for Molecular Medicine Finland Helsinki, Finland Reviewed by Professor Pirjo Laakkonen Molecular Cancer Biology Program Faculty of Medicine University of Helsinki, and A. -

Loss of the E3 Ubiquitin Ligase MKRN1 Represses Diet-Induced Metabolic Syndrome Through AMPK Activation
ARTICLE DOI: 10.1038/s41467-018-05721-4 OPEN Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation Min-Sik Lee1, Hyun-Ji Han2, Su Yeon Han2, Il Young Kim3,4, Sehyun Chae5, Choong-Sil Lee2, Sung Eun Kim2, Seul Gi Yoon4, Jun-Won Park4, Jung-Hoon Kim2, Soyeon Shin2, Manhyung Jeong2, Aram Ko2, Ho-Young Lee6, Kyoung-Jin Oh 7, Yun-Hee Lee 8, Kwang-Hee Bae7, Seung-Hoi Koo9, Jea-woo Kim10, Je Kyung Seong3,4, Daehee Hwang5 & Jaewhan Song 2 1234567890():,; AMP-activated protein kinase (AMPK) plays a key role in controlling energy metabolism in response to physiological and nutritional status. Although AMPK activation has been pro- posed as a promising molecular target for treating obesity and its related comorbidities, the use of pharmacological AMPK activators has been met with contradictory therapeutic challenges. Here we show a regulatory mechanism for AMPK through its ubiquitination and degradation by the E3 ubiquitin ligase makorin ring finger protein 1 (MKRN1). MKRN1 depletion promotes glucose consumption and suppresses lipid accumulation due to AMPK stabilisation and activation. Accordingly, MKRN1-null mice show chronic AMPK activation in both liver and adipose tissue, resulting in significant suppression of diet-induced metabolic syndrome. We demonstrate also its therapeutic effect by administering shRNA targeting MKRN1 into obese mice that reverses non-alcoholic fatty liver disease. We suggest that ubiquitin-dependent AMPK degradation represents a target therapeutic strategy for meta- bolic disorders. 1 Harvard Medical School, Boston Children’s Hospital, 3 Blackfan Circle CLS-16060.2, Boston, MA 02115, USA. 2 Department of Biochemistry, College of Life Science and Biotechnology, Yonsei University, Seoul 03722, Republic of Korea. -

PPT1 Gene Palmitoyl-Protein Thioesterase 1
PPT1 gene palmitoyl-protein thioesterase 1 Normal Function The PPT1 gene provides instructions for making an enzyme called palmitoyl-protein thioesterase 1. This enzyme is found in structures called lysosomes, which are compartments within cells that break down and recycle different types of molecules. Palmitoyl-protein thioesterase 1 removes certain fats called long-chain fatty acids from specific proteins, typically a fatty acid called palmitate. Removing these fatty acids helps break the proteins down when they are no longer needed. Palmitoyl-protein thioesterase 1 is also thought to be involved in a variety of other cell functions, such as the development of synapses, which are the connections between nerve cells where cell-to-cell communication occurs. Health Conditions Related to Genetic Changes CLN1 disease More than 65 mutations in the PPT1 gene have been found to cause CLN1 disease. This condition impairs mental and motor development causing difficulty with walking, speaking, and intellectual function. In addition, affected children often develop recurrent seizures (epilepsy) and vision loss. Signs and symptoms of CLN1 disease typically appear by age 18 months but can begin later, sometimes in adulthood, in some individuals. The PPT1 gene mutations that cause CLN1 disease decrease or eliminate the production or function of palmitoyl-protein thioesterase 1. The most common mutation causing CLN1 disease worldwide, written as Arg151Ter or R151X, replaces the protein building block (amino acid) arginine with a premature stop signal in the instructions used to make the enzyme. This mutation results in an abnormally short, nonfunctional version of the enzyme. Another mutation causes most cases of the disorder in people of Finnish descent; this genetic change replaces arginine with the amino acid tryptophan at position 122 in the palmitoyl-protein thioesterase 1 enzyme (written as Arg122Trp or R122W). -

BMC Cell Biology Biomed Central
BMC Cell Biology BioMed Central Research article Open Access Novel interactions of CLN5 support molecular networking between Neuronal Ceroid Lipofuscinosis proteins Annina Lyly†, Carina von Schantz†, Claudia Heine, Mia-Lisa Schmiedt, Tessa Sipilä, Anu Jalanko and Aija Kyttälä* Address: National Institute for Health and Welfare (THL), Biomedicum Helsinki, Finland and FIMM, Institute for Molecular Medicine in Finland Email: Annina Lyly - [email protected]; Carina von Schantz - [email protected]; Claudia Heine - [email protected]; Mia-Lisa Schmiedt - [email protected]; Tessa Sipilä - [email protected]; Anu Jalanko - [email protected]; Aija Kyttälä* - [email protected] * Corresponding author †Equal contributors Published: 26 November 2009 Received: 11 June 2009 Accepted: 26 November 2009 BMC Cell Biology 2009, 10:83 doi:10.1186/1471-2121-10-83 This article is available from: http://www.biomedcentral.com/1471-2121/10/83 © 2009 Lyly et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Neuronal ceroid lipofuscinoses (NCLs) comprise at least eight genetically characterized neurodegenerative disorders of childhood. Despite of genetic heterogeneity, the high similarity of clinical symptoms and pathology of different NCL disorders suggest cooperation between different NCL proteins and common mechanisms of pathogenesis. Here, we have studied molecular interactions between NCL proteins, concentrating specifically on the interactions of CLN5, the protein underlying the Finnish variant late infantile form of NCL (vLINCLFin). -

The Phosphopantetheinyl Transferases: Catalysis of a Post-Translational Modification Crucial for Life† Cite This: Nat
NPR REVIEW View Article Online View Journal | View Issue The phosphopantetheinyl transferases: catalysis of a post-translational modification crucial for life† Cite this: Nat. Prod. Rep.,2014,31,61 Joris Beld,‡a Eva C. Sonnenschein,‡§a Christopher R. Vickery,‡ab Joseph P. Noelb and Michael D. Burkart*a Covering: up to 2013 Although holo-acyl carrier protein synthase, AcpS, a phosphopantetheinyl transferase (PPTase), was characterized in the 1960s, it was not until the publication of the landmark paper by Lambalot et al. in 1996 that PPTases garnered wide-spread attention being classified as a distinct enzyme superfamily. In the past two decades an increasing number of papers have been published on PPTases ranging from Received 11th June 2013 identification, characterization, structure determination, mutagenesis, inhibition, and engineering in DOI: 10.1039/c3np70054b synthetic biology. In this review, we comprehensively discuss all current knowledge on this class of www.rsc.org/npr enzymes that post-translationally install a 40-phosphopantetheine arm on various carrier proteins. 1 Introduction 4.2 The other phosphopantetheinylated proteins and their 2 Types of PPTases PPTases 2.1 Family I: holo-acyl carrier protein synthase (AcpS-type 4.3 Carrier protein recognition by PPTases PPTases) 4.4 Peptide mimics of carrier proteins as substrate of PPTases 2.2 Family II: Sfp-type PPTases 4.5 Regulation by 40-phosphopantetheinylation 2.3 Family III: type I integrated PPTases 5 Structures 3 Importance in primary and secondary metabolism 5.1 Structural -
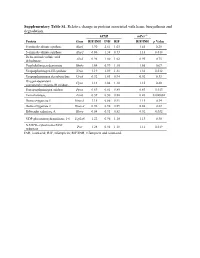
Supplementary Table S1. Relative Change in Proteins Associated with Heme Biosynthesis and Degradation
Supplementary Table S1. Relative change in proteins associated with heme biosynthesis and degradation. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value 5-aminolevulinate synthase Alas1 1.90 2.61 1.05 1.41 0.28 5-aminolevulinate synthase Alas2 0.86 1.38 0.73 1.18 0.018 Delta-aminolevulinic acid Alad 0.96 1.00 1.02 0.95 0.75 dehydratase Porphobilinogen deaminase Hmbs 1.04 0.99 1.10 1.05 0.67 Uroporphyrinogen-III synthase Uros 1.19 1.09 1.31 1.38 0.012 Uroporphyrinogen decarboxylase Urod 0.92 1.03 0.94 0.92 0.33 Oxygen-dependent Cpox 1.13 1.04 1.18 1.15 0.20 coproporphyrinogen-III oxidase, Protoporphyrinogen oxidase Ppox 0.69 0.81 0.85 0.83 0.013 Ferrochelatase, Fech 0.39 0.50 0.88 0.43 0.000002 Heme oxygenase 1 Hmox1 1.15 0.86 0.91 1.11 0.34 Heme oxygenase 2 Hmox2 0.96 0.98 0.89 0.88 0.22 Biliverdin reductase A Blvra 0.84 0.92 0.82 0.92 0.032 UDP-glucuronosyltransferase 1-6 Ugt1a6 1.22 0.96 1.10 1.13 0.30 NADPH--cytochrome P450 Por 1.28 0.92 1.18 1.12 0.019 reductase INH, isoniazid; RIF, rifampicin; RIF/INH, rifampicin and isoniazid. Supplementary Table S2. Relative change in protein nuclear receptors. hPXR mPxr–/– Protein Gene RIF/INH INH RIF RIF/INH p Value Aryl hydrocarbon receptor Ahr 1.09 0.91 1.00 1.26 0.092 Hepatocyte nuclear factor Hnf1a 0.87 0.97 0.82 0.79 0.027 1-alpha Hepatocyte nuclear factor Hnf4a 0.95 1.05 0.97 1.08 0.20 4-alpha Oxysterols receptor LXR- Nr1h3 0.94 1.16 1.03 1.02 0.42 alpha Bile acid receptor Nr1h4 1.05 1.17 0.98 1.19 0.12 Retinoic acid receptor Rxra 0.88 1.03 0.83 0.95 0.12 RXR-alpha Peroxisome proliferator- -

The Crystal Structure of Palmitoyl Protein Thioesterase 1 and the Molecular Basis of Infantile Neuronal Ceroid Lipofuscinosis
The crystal structure of palmitoyl protein thioesterase 1 and the molecular basis of infantile neuronal ceroid lipofuscinosis John J. Bellizzi III*, Joanne Widom*, Christopher Kemp†, Jui-Yun Lu‡, Amit K. Das‡, Sandra L. Hofmann‡, and Jon Clardy*§ *Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853; †Kemp Biotechnologies, Frederick, MD 21704; and ‡Department of Internal Medicine and the Hamon Center for Therapeutic Oncology Research, University of Texas Southwestern Medical Center, Dallas, TX 75235 Edited by William N. Lipscomb, Harvard University, Cambridge, MA, and approved February 14, 2000 (received for review November 22, 1999) Mutations in palmitoyl-protein thioesterase 1 (PPT1), a lysosomal represents the only eukaryotic thioesterase to be examined at enzyme that removes fatty acyl groups from cysteine residues in atomic resolution. modified proteins, cause the fatal inherited neurodegenerative We determined the crystal structure of bovine PPT1 by using disorder infantile neuronal ceroid lipofuscinosis. The accumulation the multiwavelength anomalous diffraction phasing method on of undigested substrates leads to the formation of neuronal a crystal of selenomethionine (SeMet) labeled PPT1. Bovine storage bodies that are associated with the clinical symptoms. Less PPT1 is 95% identical to human PPT1 at the amino acid level severe forms of PPT1 deficiency have been found recently that are (14). The structure has been refined to 2.25 Å resolution. We caused by a distinct set of PPT1 mutations, some of which retain a have also determined the structure of a covalent acyl-enzyme small amount of thioesterase activity. We have determined the complex between PPT1 and palmitate, which has been refined crystal structure of PPT1 with and without bound palmitate by to 2.5 Å resolution.