Iron Chelation Inhibits Mtorc1 Signaling Involving Activation of AMPK and REDD1/Bnip3 Pathways
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Autophagy: from Basic Science to Clinical Application
nature publishing group REVIEW See COMMENTARY page XX Autophagy: from basic science to clinical application J Va n L i m b e r g e n 1 , 2 , 3 , C S t e v e n s 4 , E R N i m m o 1 , D C W i l s o n 2 , 3 a n d J S a t s a n g i 1 Autophagy is a cellular pathway involved in protein and organelle degradation, which is likely to represent an innate adaptation to starvation. In times of nutrient deficiency, the cell can self-digest and recycle some nonessential components through nonselective autophagy, thus sustaining minimal growth requirements until a food source becomes available. Over recent years, autophagy has been implicated in an increasing number of clinical scenarios, notably infectious diseases, cancer, neurodegenerative diseases, and autoimmunity. The recent identification of the importance of autophagy genes in the genetic susceptibility to Crohn ’ s disease suggests that a selective autophagic response may play a crucial role in the pathogenesis of common complex immune-mediated diseases. In this review, we discuss the autophagic mechanisms, their molecular regulation, and summarize their clinical relevance. This progress has led to great interest in the therapeutic potential of manipulation of both selective and nonselective autophagy in established disease. INTRODUCTION The ability to adapt to environmental change is essential for sur- Autophagy encompasses several distinct processes involving vival. This is true for the organism as a whole and for individual the delivery of portions of the cytoplasm to the lysosome for cells alike. -

The Retinoblastoma Tumor-Suppressor Gene, the Exception That Proves the Rule
Oncogene (2006) 25, 5233–5243 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc REVIEW The retinoblastoma tumor-suppressor gene, the exception that proves the rule DW Goodrich Department of Pharmacology & Therapeutics, Roswell Park Cancer Institute, Buffalo, NY, USA The retinoblastoma tumor-suppressor gene (Rb1)is transmission of one mutationally inactivated Rb1 allele centrally important in cancer research. Mutational and loss of the remaining wild-type allele in somatic inactivation of Rb1 causes the pediatric cancer retino- retinal cells. Hence hereditary retinoblastoma typically blastoma, while deregulation ofthe pathway in which it has an earlier onset and a greater number of tumor foci functions is common in most types of human cancer. The than sporadic retinoblastoma where both Rb1 alleles Rb1-encoded protein (pRb) is well known as a general cell must be inactivated in somatic retinal cells. To this day, cycle regulator, and this activity is critical for pRb- Rb1 remains an exception among cancer-associated mediated tumor suppression. The main focus of this genes in that its mutation is apparently both necessary review, however, is on more recent evidence demonstrating and sufficient, or at least rate limiting, for the genesis of the existence ofadditional, cell type-specific pRb func- a human cancer. The simple genetics of retinoblastoma tions in cellular differentiation and survival. These has spawned the hope that a complete molecular additional functions are relevant to carcinogenesis sug- understanding of the Rb1-encoded protein (pRb) would gesting that the net effect of Rb1 loss on the behavior of lead to deeper insight into the processes of neoplastic resulting tumors is highly dependent on biological context. -

Central Nervous System Cancers Panel Members Can Be Found on Page 1151
1114 NCCN David Tran, MD, PhD; Nam Tran, MD, PhD; Frank D. Vrionis, MD, MPH, PhD; Patrick Y. Wen, MD; Central Nervous Nicole McMillian, MS; and Maria Ho, PhD System Cancers Overview In 2013, an estimated 23,130 people in the United Clinical Practice Guidelines in Oncology States will be diagnosed with primary malignant brain Louis Burt Nabors, MD; Mario Ammirati, MD, MBA; and other central nervous system (CNS) neoplasms.1 Philip J. Bierman, MD; Henry Brem, MD; Nicholas Butowski, MD; These tumors will be responsible for approximately Marc C. Chamberlain, MD; Lisa M. DeAngelis, MD; 14,080 deaths. The incidence of primary brain tumors Robert A. Fenstermaker, MD; Allan Friedman, MD; Mark R. Gilbert, MD; Deneen Hesser, MSHSA, RN, OCN; has been increasing over the past 30 years, especially in Matthias Holdhoff, MD, PhD; Larry Junck, MD; elderly persons.2 Metastatic disease to the CNS occurs Ronald Lawson, MD; Jay S. Loeffler, MD; Moshe H. Maor, MD; much more frequently, with an estimated incidence ap- Paul L. Moots, MD; Tara Morrison, MD; proximately 10 times that of primary brain tumors. An Maciej M. Mrugala, MD, PhD, MPH; Herbert B. Newton, MD; Jana Portnow, MD; Jeffrey J. Raizer, MD; Lawrence Recht, MD; estimated 20% to 40% of patients with systemic cancer Dennis C. Shrieve, MD, PhD; Allen K. Sills Jr, MD; will develop brain metastases.3 Abstract Please Note Primary and metastatic tumors of the central nervous system are The NCCN Clinical Practice Guidelines in Oncology a heterogeneous group of neoplasms with varied outcomes and (NCCN Guidelines®) are a statement of consensus of the management strategies. -

Characterization of Gf a Drosophila Trimeric G Protein Alpha Subunit
Characterization of Gf a Drosophila trimeric G protein alpha subunit Naureen Quibria Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2012 © 2012 Naureen Quibria All rights reserved Abstract Characterization of Gf a Drosophila trimeric G-protein alpha subunit Naureen Quibria In the morphogenesis of tissue development, how coordination of patterning and growth achieve the correct organ size and shape is a principal question in biology. Efficient orchestrating mechanisms are required to achieve this and cells have developed sophisticated systems for reception and interpretation of the multitude of extracellular stimuli to which they are exposed. Plasma membrane receptors play a key role in the transmission of such signals. G-protein coupled receptors (GPCRs) are the largest class of cell surface receptors that respond to an enormous diversity of extracellular stimuli, and are critical mediators of cellular signal transduction in eukaryotic organisms. Signaling through GPCRs has been well characterized in many biological contexts. While they are a major class of signal transducers, there are not many defined instances where GPCRs have been implicated in the process of development to date. The Drosophila wing provides an ideal model system to elucidate and address the role of GPCRs in development, as its growth is regulated by a small number of conserved signaling pathways. In my thesis work, I address the role of a trimeric G alpha protein in Drosophila, Gαf, and what part it may play in development. In particular, I explore the role of Gαf as an alpha subunit of a trimeric complex, to determine what heptahelical receptors might act as its cognate receptor. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genetics of Tuberous Sclerosis Complex: Implications for Clinical Practice
Journal name: The Application of Clinical Genetics Article Designation: REVIEW Year: 2017 Volume: 10 The Application of Clinical Genetics Dovepress Running head verso: Caban et al Running head recto: Genetics of TSC open access to scientific and medical research DOI: http://dx.doi.org/10.2147/TACG.S90262 Open Access Full Text Article REVIEW Genetics of tuberous sclerosis complex: implications for clinical practice Carolina Caban1,2 Abstract: Tuberous sclerosis complex (TSC) is a multisystem disorder that results from hetero- Nubaira Khan1,2 zygous mutations in either TSC1 or TSC2. The primary organ systems that are affected include Daphne M Hasbani3 the brain, skin, lung, kidney, and heart, all with variable frequency, penetrance, and severity. Peter B Crino1,2 Neurological features include epilepsy, autism, and intellectual disability. There are more than 1,500 known pathogenic variants for TSC1 and TSC2, including deletion, nonsense, and missense 1Department of Neurology, 2Shriners Hospitals Pediatric Research mutations, and all pathogenic mutations are inactivating, leading to loss of function effects on Center, Temple University School of the encoded proteins TSC1 and TSC2. These proteins form a complex to constitutively inhibit 3 Medicine, Department of Neurology, mechanistic target of rapamycin (mTOR) signaling cascade, and as a consequence, mTOR signal- St. Christopher’s Hospital for Children, Drexel University College ing is constitutively active within all TSC-associated lesions. The mTOR inhibitors rapamycin For personal use only. of Medicine, Philadelphia, PA, USA (sirolimus) and everolimus have been shown to reduce the size of renal and brain lesions and improve pulmonary function in TSC, and these compounds may also decrease seizure frequency. -

Inhibitors of Mammalian Target of Rapamycin Downregulate MYCN Protein Expression and Inhibit Neuroblastoma Growth in Vitro and in Vivo
Oncogene (2008) 27, 2910–2922 & 2008 Nature Publishing Group All rights reserved 0950-9232/08 $30.00 www.nature.com/onc ORIGINAL ARTICLE Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo JI Johnsen1,6, L Segerstro¨ m1,6, A Orrego2, L Elfman1, M Henriksson3,BKa˚ gedal4, S Eksborg1, B Sveinbjo¨ rnsson1,5 and P Kogner1 1Department of Woman and Child Health, Karolinska Institutet, Childhood Cancer Research Unit, Stockholm, Sweden; 2Department of Oncology and Pathology, Karolinska Institutet, Stockholm, Sweden; 3Department of Microbiology, Tumor and Cellbiology, Karolinska Institutet, Stockholm, Sweden; 4Division of Clinical Chemistry, Faculty of Health Sciences, Linko¨ping University, Sweden and 5Department of Cell Biology and Histology, University of Tromso¨, Tromso¨, Norway Mammalian target of rapamycin (mTOR) has been shown the most common and deadly solid tumor of childhood to play an important function in cell proliferation, (Brodeur, 2003). Amplification of the MYCN oncogene metabolism and tumorigenesis, and proteins that regulate is associated with rapid tumor progression and fre- signaling through mTOR are frequently altered in human quently detected in advanced-stage neuroblastoma, but cancers. In this study we investigated the phosphorylation is also a major negative prognostic factor in localized status of key proteins in the PI3K/AKT/mTOR pathway tumors (Schwab et al., 2003). Advanced-stage tumors and the effects of the mTOR inhibitors rapamycin and and those with MYCN amplification show typically CCI-779 on neuroblastoma tumorigenesis. Significant emergence of treatment resistance, and alternative expression of activated AKT and mTOR were detected treatment strategies for these patients are therefore in all primary neuroblastoma tissue samples investigated, urgently needed. -
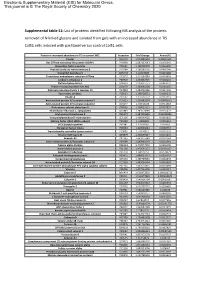
Supplemental Table S1
Electronic Supplementary Material (ESI) for Molecular Omics. This journal is © The Royal Society of Chemistry 2020 Supplemental table S1: List of proteins identified following MS analysis of the proteins removed of N-linked glycans and isolated from gels with an increased abundance in TIS Cal51 cells induced with paclitaxel versus control Cal51 cells. Protein in increased abundance in TIS vs control WCL Accession Fold Change Anova (P) Plectin Q15149 1.073855593 0.00691631 Ras GTPase-activating-like protein IQGAP1 P46940 1.087337643 0.0176342 Elongation factor1-gamma P26641 1.138709703 0.0116496 Peptidyl-prolyl cis-transisomerase B P23284 1.188383105 0.0436246 Dipeptidyl peptidase 3 Q9NY33 1.20163605 0.0215448 Transitional endoplasmic reticulum ATPase P55072 1.214194884 0.0449691 Carbonic anhydrase 2 P00918 1.232852325 0.0158141 Clathrin heavy chain 1 Q00610 1.239621773 0.0463237 Protein transport protein Sec 31A O94979 1.263565104 0.0284155 Aldo-ketoreductase family 1 member C1 Q04828 1.282092186 0.0324406 Spermidine synthase P19623 1.298728621 0.0196232 Plastin-3 P13797 1.310756772 0.0161319 Actin-related protein 2/3 complex subunit 5 O15511 1.333483524 0.00476923 Actin-related protein 2/3 complex subunit 2 O15144 1.35416168 0.0411018 Proteasome subunit alpha type-5 P28066 1.358015551 0.0337657 Thioredoxin reductase 1, cytoplasmic Q16881 1.383670089 0.0235472 Acyl-protein thioesterase 2 O95372 1.387415589 0.00233899 Isoaspartylpeptidase/L-asparaginase Q7L266 1.408149002 0.0319602 Splicing factor U2AF 65kDa subunit P26368 1.41489991 0.0256619 -

Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/Mtor Cascade Inhibitors: How Mutations Can Result in Therapy Resistance and How to Overcome Resistance
www.impactjournals.com/oncotarget/ Oncotarget, October, Vol.3, No 10 Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascade Inhibitors: How Mutations Can Result in Therapy Resistance and How to Overcome Resistance James A. McCubrey1, Linda S. Steelman1, William H. Chappell1, Stephen L. Abrams1, Richard A. Franklin1, Giuseppe Montalto2, Melchiorre Cervello3, Massimo Libra4, Saverio Candido4, Grazia Malaponte4, Maria C. Mazzarino4, Paolo Fagone4, Ferdinando Nicoletti4, Jörg Bäsecke5, Sanja Mijatovic6, Danijela Maksimovic- Ivanic6, Michele Milella7, Agostino Tafuri8, Francesca Chiarini9, Camilla Evangelisti9, Lucio Cocco10, Alberto M. Martelli9,10 1 Department of Microbiology and Immunology, Brody School of Medicine at East Carolina University, Greenville, NC, USA 2 Department of Internal Medicine and Specialties, University of Palermo, Palermo, Italy 3 Consiglio Nazionale delle Ricerche, Istituto di Biomedicina e Immunologia Molecolare “Alberto Monroy”, Palermo, Italy 4 Department of Bio-Medical Sciences, University of Catania, Catania, Italy 5 Department of Medicine, University of Göttingen, Göttingen, Germany 6 Department of Immunology, Instititue for Biological Research “Sinisa Stankovic”, University of Belgrade, Belgrade, Serbia 7 Regina Elena National Cancer Institute, Rome, Italy 8 Sapienza, University of Rome, Department of Cellular Biotechnology and Hematology, Rome, Italy 9 Institute of Molecular Genetics, National Research Council-Rizzoli Orthopedic Institute, Bologna, Italy. 10 Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy Correspondence to: James A. McCubrey, email: [email protected] Keywords: Targeted Therapy, Therapy Resistance, Cancer Stem Cells, Raf, Akt, PI3K, mTOR Received: September 12, 2012, Accepted: October 18, 2012, Published: October 20, 2012 Copyright: © McCubrey et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. -

1P36 Tumor Suppression—A Matter of Dosage?
Published OnlineFirst November 20, 2012; DOI: 10.1158/0008-5472.CAN-12-2230 Cancer Review Research 1p36 Tumor Suppression—A Matter of Dosage? Kai-Oliver Henrich, Manfred Schwab, and Frank Westermann Abstract A broad range of human malignancies is associated with nonrandom 1p36 deletions, suggesting the existence of tumor suppressors encoded in this region. Evidence for tumor-specific inactivation of 1p36 genes in the classic "two-hit" manner is scarce; however, many tumor suppressors do not require complete inactivation but contribute to tumorigenesis by partial impairment. We discuss recent data derived from both human tumors and functional cancer models indicating that the 1p36 genes CHD5, CAMTA1, KIF1B, CASZ1, and miR-34a contribute to cancer development when reduced in dosage by genomic copy number loss or other mechanisms. We explore potential interactions among these candidates and propose a model where heterozygous 1p36 deletion impairs oncosuppressive pathways via simultaneous downregulation of several dosage-dependent tumor suppressor genes. Cancer Res; 72(23); 1–10. Ó2012 AACR. Introduction (Fig. 1; refs. 1, 17–29). Despite extensive 1p36 candidate gene Deletions of the distal short arm of chromosome 1 (1p) are sequence analyses, success was limited for identifying tumor- fi frequently observed in a broad range of human cancers, speci c mutations in neuroblastomas or other malignancies, including breast cancer, cervical cancer, pancreatic cancer, which led some to conclude that a deletion mapping approach pheochromocytoma, thyroid cancer, hepatocellular cancer, was unlikely to deliver tumor suppressor genes. Many tumor colorectal cancer, lung cancer, glioma, meningioma, neuro- suppressor genes, however, do not require inactivation in a blastoma, melanoma, Merkel cell carcinoma, rhabdomyosar- classic "two-hit" manner but contribute to tumor development coma, acute myeloid leukemia, chronic myeloid leukemia, and when their dosage is reduced, sometimes only subtly, by non-Hodgkin lymphoma (1, 2). -

Melanomas Are Comprised of Multiple Biologically Distinct Categories
Melanomas are comprised of multiple biologically distinct categories, which differ in cell of origin, age of onset, clinical and histologic presentation, pattern of metastasis, ethnic distribution, causative role of UV radiation, predisposing germ line alterations, mutational processes, and patterns of somatic mutations. Neoplasms are initiated by gain of function mutations in one of several primary oncogenes, typically leading to benign melanocytic nevi with characteristic histologic features. The progression of nevi is restrained by multiple tumor suppressive mechanisms. Secondary genetic alterations override these barriers and promote intermediate or overtly malignant tumors along distinct progression trajectories. The current knowledge about pathogenesis, clinical, histological and genetic features of primary melanocytic neoplasms is reviewed and integrated into a taxonomic framework. THE MOLECULAR PATHOLOGY OF MELANOMA: AN INTEGRATED TAXONOMY OF MELANOCYTIC NEOPLASIA Boris C. Bastian Corresponding Author: Boris C. Bastian, M.D. Ph.D. Gerson & Barbara Bass Bakar Distinguished Professor of Cancer Biology Departments of Dermatology and Pathology University of California, San Francisco UCSF Cardiovascular Research Institute 555 Mission Bay Blvd South Box 3118, Room 252K San Francisco, CA 94158-9001 [email protected] Key words: Genetics Pathogenesis Classification Mutation Nevi Table of Contents Molecular pathogenesis of melanocytic neoplasia .................................................... 1 Classification of melanocytic neoplasms -

Molecular Docking and Pharmacokinetic of Highly Specific Novel Pan-Mtor Inhibitors Against Solid Tumors
MOJ Proteomics & Bioinformatics Research Article Open Access Molecular docking and pharmacokinetic of highly specific novel pan-mtor inhibitors against solid tumors Abstract Volume 5 Issue 6 - 2017 Mechanistic/mammalian target of rapamycin (mTOR) a serine/threonine kinase belonging Muhammad Naveed,1,2 Safia Zia,1 Maryam to PI3K/Akt/mTOR pathway is involved in different cellular functions cell survival, 1 1 1 metabolism, growth, proliferation, apoptosis and autophagy. Pan-mTOR inhibitors are Akhtar, Fatima Ashraf, Amber Afroz 1Department of Biochemistry and Biotechnology, University of targeted towards mTOR dysregulation, inhibiting the kinase domain of both mTORC1 Gujrat, Pakistan and mTORC2. The present study analyzes the binding modes and molecular interactions 2Department of Biotechnology, University of Gujrat, Pakistan of highly specific mTOR inhibitors, AZD8055 and its sister compoundAZD2014using computational approach. Both inhibitors proved to be effective against solid tumors in Correspondence: Muhammad Naveed, Department of vitro and in vivo. Docking analysis was performed using Auto Dock Vina, conformations Biochemistry and Biotechnology, University of Gujrat, Pakistan, were scored based upon their binding energy (kcal/mol) and illustrated using Discovery Tel 00923015524624, Email [email protected] Studio Visualizer 4.5 version. Inhibitors fit between N- and C-lobes of mTOR kinase domain into the inner hydrophobic core. The results indicated interactions with distinctive Received: February 03, 2017 | Published: June 27, 2017 mTOR residues Trp-2239, Leu-2185 and newly developed interactions with Asp-2375 for AZD2014 and with Ala-2248, His-2247, Thr-2245 for AZD8055. The binding pattern of both inhibitors was slightly different, responsible for better pharmacokinetic profile of AZD2014 and 5 fold increase in efficacy of AZD8055.