Charlotte L. Mattsson
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Keith Dougherty
KEITH DOUGHERTY Office Department of Political Science updated 10/15/20 University of Georgia [email protected] Athens, Georgia 30602 Education Ph.D. 1997. Government and Politics. University of Maryland. M.A. 1992. Government and Politics. University of Maryland. B.A. 1988. Political Economy. Tulane University. Positions Professor, University of Georgia, Athens, Georgia, 2014-present. Associate Professor, University of Georgia, Athens, Georgia, 2006-2014. Visiting Professor, Social and Decision Sciences, Carnegie Mellon University, Pittsburgh, Pennsylvania, spring 2006. Assistant Professor, University of Georgia, Athens, Georgia, 2003-2006. Assistant Professor, Florida International University, Miami, Florida, 1999-2003. Visiting Scholar, The Workshop in Political Theory and Policy Analysis, Indiana University, Bloomington, Indiana, 1998-1999. Visiting Professor, St. Mary’s College of Maryland, St. Mary’s City, Maryland, 1997-1998. Editorial Associate Editor (Political Science), Public Choice, 2016 - 2019. Position Grants National Science Foundation, SES-1154920 ($22,963) for “The American Constitution: A Conference on the 225th Anniversary of the Ratification,” with Keith Poole and Peter Hoffer, 2012-2013. National Science Foundation, SES-0752098 ($205,000) for “Delegate Voting at the Constitutional Convention,” with Jac Heckelman, 2008-2010. National Science Foundation, SES-0418254 ($71,000) for “American Founding: Motivations of the Framers at the Constitutional Convention,” with Jac Heckelman, 2004-2005. Fellowships Lothar Tresp Outstanding Honors Professor Award, Honors College, 2016. and Awards Teaching Award, School of Public and International Affairs, 2015. Outstanding Professor Award, UGA Student Government Association, 2014. Research Award, School of Public and International Affairs, 2014. Gordon Tullock Prize for best article in Public Choice by younger scholars, 2008. Fulbright Scholarship, Tomsk State University, Russia, 1998-1999 (declined). -

A. Cruise Narrative: AR25 A.1. Highlights WHP Cruise
A. Cruise Narrative: AR25 .565 .555 .538 .520 A.1. Highlights WHP Cruise Summary Information WOCE section designation AR25 Expedition designation (EXPOCODE) 06MT45_4 Chief Scientist/affiliation Jens Meincke/IfMH* Dates 1999 AUG 13 - 1999 AUG 31 Ship R/V METEOR Ports of call St. John's, Newfoundland – Rendsburg, Germany Number of stations 46 65°31.19’N Stations’ geographic boundaries 49°22.91’W 27°14.59’W 51°54.77’N Floats and drifters deployed see section 4.2; LEG 2 Moorings deployed or recovered 8 recovered and redeployed Contributing Authors J. Meincke O. Plähn K. Bulsiewicz I. Schlimme G. Kahl *Institut für Meereskunde an der Universität Hamburg • Troplowitzstraße 7 22529 Hamburg • GERMANY • TEL: +49-40-4123-5985 • FAX: +49-40-4123-4644 EMAIL: [email protected] WHP Cruise and Data Information Instructions: Click on headings below to locate primary reference or use navigation tools above. (Shaded headings were not available when this report was assembled) Cruise Summary Information Hydrographic Measurements Description of scientific program CTD Data Geographic boundaries of the survey CTD - general Cruise track (WHPO) (PI) CTD - pressure Description of stations CTD - temperature Description of parameters sampled CTD - conductivity/salinity Bottle depth distributions (figure) CTD - dissolved oxygen Floats and drifters deployed Moorings deployed or recovered Bottle Data Salinity Principal Investigators for all measurements Oxygen Cruise Participants Nutrients CFCs Problems and goals not achieved Helium Other incidents of -

Carlson 5 Report
STATE OF ALASKA Commercial Fisheries Entry Commission Carlson Summary Only those with a Positive Balance as of 31 January 2010 Page 1 of 91 Non-Resident Over/Under Balance Fee Differential Interest as of Name Paid Allowance Amount 31Jan2010 Obs —————————————————————————————— —————————————— —————————————— —————————————— —————————————— 1 AADLAND, ARNE $15,750.00 $7,993.37 $64,755.64 $72,749.01 2 AAKER, MERLE O. $7,110.00 $1,466.09 $8,111.91 $9,578.00 3 AALMO, ARNOLD $1,500.00 $273.38 $745.70 $1,019.08 4 AARVIK, STEVE $12,685.00 $4,243.93 $18,625.51 $22,869.44 5 AASE, CARL E. (ESTATE) $2,005.00 $166.56 $363.10 $529.66 6 AASE, OLAF H. (ESTATE) $8,700.00 $2,720.71 $17,078.65 $19,799.36 7 ABBOTT, RICHARD $450.00 $84.03 $795.01 $879.04 8 ABBOTT, THOMAS $6,295.00 $2,137.46 $9,588.06 $11,725.52 9 ABE, TOSHINARI $990.00 $150.08 $928.56 $1,078.64 10 ABRAHAM, BRUCE E. $1,590.00 $306.69 $2,309.41 $2,616.10 11 ACCETTA, MATTEO $7,350.00 $2,393.37 $16,841.25 $19,234.62 12 ACCOUSTI, NEAL O. $900.00 $62.66 $526.77 $589.43 13 ACKERMANN, RANDY D. $9,390.00 $3,943.99 $21,771.75 $25,715.74 14 ADAMS, CHARLES F. Jr. $12,415.00 $3,978.30 $21,420.42 $25,398.72 15 ADAMS, DALE $1,720.00 $190.06 $4,821.80 $5,011.86 16 ADAMS, DEAN J. -
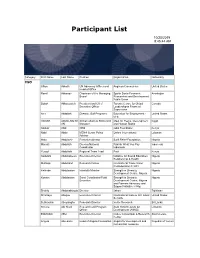
Participant List
Participant List 10/20/2019 8:45:44 AM Category First Name Last Name Position Organization Nationality CSO Jillian Abballe UN Advocacy Officer and Anglican Communion United States Head of Office Ramil Abbasov Chariman of the Managing Spektr Socio-Economic Azerbaijan Board Researches and Development Public Union Babak Abbaszadeh President and Chief Toronto Centre for Global Canada Executive Officer Leadership in Financial Supervision Amr Abdallah Director, Gulf Programs Educaiton for Employment - United States EFE HAGAR ABDELRAHM African affairs & SDGs Unit Maat for Peace, Development Egypt AN Manager and Human Rights Abukar Abdi CEO Juba Foundation Kenya Nabil Abdo MENA Senior Policy Oxfam International Lebanon Advisor Mala Abdulaziz Executive director Swift Relief Foundation Nigeria Maryati Abdullah Director/National Publish What You Pay Indonesia Coordinator Indonesia Yussuf Abdullahi Regional Team Lead Pact Kenya Abdulahi Abdulraheem Executive Director Initiative for Sound Education Nigeria Relationship & Health Muttaqa Abdulra'uf Research Fellow International Trade Union Nigeria Confederation (ITUC) Kehinde Abdulsalam Interfaith Minister Strength in Diversity Nigeria Development Centre, Nigeria Kassim Abdulsalam Zonal Coordinator/Field Strength in Diversity Nigeria Executive Development Centre, Nigeria and Farmers Advocacy and Support Initiative in Nig Shahlo Abdunabizoda Director Jahon Tajikistan Shontaye Abegaz Executive Director International Insitute for Human United States Security Subhashini Abeysinghe Research Director Verite -

Graduating Undergraduates with Concentration at Which Receiving Diploma Spring 2016
Graduating Undergraduates with Concentration at Which Receiving Diploma Spring 2016 Name Degree Concentration Departmental Ceremony Abarca, Cindy Jasmin AB Public Health Public Health Abdel-Haq, Reem Ali SCB Biology Biology Abe, Tathya Yuki AB Environmental Studies Environ Sci & Environ Studies Abeygunawardana, Melanie Enoka AB English English Abu-Akeel, Tariq Shereef AB Psychology Cognitv, Ling, & Psych Sciences Acero, Darien Rafael Besteman AB Religious Studies Religious Studies Ackmann, Christian Michael AB Economics Economics Acosta, Kailani Gabriella SCB Environmental Science Environ Sci & Environ Studies Adam, Jonathan AB Music Music Adams, Chanelle AB Science and Society Science and Society Adams, Dominic Tedeschi SCB Physics Physics Adams, Kyle William AB History History Addy, Jason AB Education Studies Educational Studies Adegbulugbe, Esther Iyiadepo SCB Cognitive Neuroscience Cognitv, Ling, & Psych Sciences Adler, Grace Elizabeth AB Literary Arts Literary Arts Adler, John Charles SCB Computer Science Computer Science Aguiar, Katherine Amanda SCB Engineering Engineering Aguilar, Zachary Penati SCB Neuroscience Neuroscience Ahiligwo, Jonelle Uzoamaka AB Public Health Public Health Ahn, Lawrence Byungkyu AB Biology Biology Ainsworth, Samuel Kenneth SCB Applied Math.-Computer Sci. Computer Science Akande, Morayo Ominira AB Public Health Public Health Akinkugbe, Oludolapo Iroloye AB Classics Classics Akinsulire, Olajumoke Mary AB Biology Biology Akram-Boshar, Shireen Najla AB Africana Studies Africana Studies Al-Eryani, Raba Abdulelah -

Last Name First Name Birth Yrfather's Last Name Father's
Last Name First Name Birth YrFather's Last Name Father's First Name Mother's Last Name Mother's First Name Gender Aaron 1907 Aaron Benjaman Shields Minnie M Aaslund 1893 Aaslund Ole Johnson Augusta M Aaslund (Twins) 1895 Aaslund Oluf Carlson Augusta F Abbeal 1906 Abbeal William A Conlee Nina E M Abbitz Bertha Dora 1896 Abbitz Albert Keller Caroline F Abbot 1905 Abbot Earl R Seldorff Rose F Abbott Zella 1891 Abbott James H Perry Lissie F Abbott 1896 Abbott Marion Forder Charolotta M M Abbott 1904 Abbott Earl R Van Horn Rose M Abbott 1906 Abbott Earl R Silsdorff Rose M Abercrombie 1899 Abercrombie W Rogers A F Abernathy Marjorie May 1907 Abernathy Elmer Scott Margaret F Abernathy 1892 Abernathy Wm A Roberts Laura J F Abernethy 1905 Abernethy Elmer R Scott Margaret M M Abikz Louisa E 1902 Abikz Albert Keller Caroline F Abilz Charles 1903 Abilz Albert Keller Caroline M Abircombie 1901 Abircombie W A Racher Allos F Abitz Arthur Carl 1899 Abitz Albert Keller Caroline M Abrams 1902 Abrams L E Baker May F Absher 1905 Absher Ben Spillman Zoida M Achermann Bernadine W 1904 Ackermann Arthur Krone Karolina F Acker 1903 Acker Louis Carr Lena F Acker 1907 Acker Leyland Ryan Beatrice M Ackerman 1904 Ackerman Cecil Addison Willis Bessie May F Ackermann Berwyn 1905 Ackermann Max Mann Dolly M Acklengton 1892 Achlengton A A Riacting Nattie F Acton Rebecca Elizabeth 1891 Acton T M Cox Josie E F Acton 1900 Acton Chas Payne Minnie M Adair Elles 1906 Adair Adel M Last Name First Name Birth YrFather's Last Name Father's First Name Mother's Last Name Mother's -
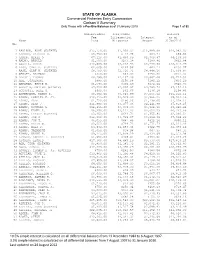
Carlson Exhibit Number 1
STATE OF ALASKA Commercial Fisheries Entry Commission Carlson V Summary Only Those with a Positive Balance as of 31January 2010 Page 1 of 92 Non-Resident Over/Under Balance Fee Differential Interest as of Name Paid Allowance Amount 31Jan2010 Obs —————————————————————————————— —————————————— —————————————— —————————————— —————————————— 1 AADLAND, ARNE (ESTATE) $15,750.00 $7,993.37 $16,989.68 $24,983.05 2 AADSEN, VALERIE S. $3,750.00 $-24.78 $84.71 $59.93 3 AAKER, MERLE O. $7,110.00 $1,466.09 $2,669.87 $4,135.96 4 AALMO, ARNOLD $1,500.00 $273.38 $189.46 $462.84 5 AARVIK, STEVE $12,685.00 $4,243.93 $5,770.86 $10,014.79 6 AASE, CARL E. (ESTATE) $2,005.00 $166.56 $64.21 $230.77 7 AASE, OLAF H. (ESTATE) $8,700.00 $2,720.71 $4,844.16 $7,564.87 8 ABBOTT, RICHARD $450.00 $84.03 $193.32 $277.35 9 ABBOTT, THOMAS $6,295.00 $2,137.46 $3,587.08 $5,724.54 10 ABE, TOSHINARI $990.00 $150.08 $286.12 $436.20 11 ABRAHAM, BRUCE E. $1,590.00 $306.69 $634.26 $940.95 12 ACCETTA, MATTEO (ESTATE) $7,350.00 $2,393.37 $4,760.73 $7,154.10 13 ACCOUSTI, NEAL O. $900.00 $62.66 $136.18 $198.84 14 ACKERMANN, RANDY D. $9,390.00 $3,943.99 $7,066.12 $11,010.11 15 ADAMS, CHARLES F. Jr. $12,415.00 $3,978.30 $5,966.26 $9,944.56 16 ADAMS, DALE $1,720.00 $190.06 $1,138.05 $1,328.11 17 ADAMS, DEAN J. -

Marriages 1884884 -- 19201930 Groom Last Name Groom First Name Bride Last Name Bride First Name Marriage Year Aadland Nels Paulson Lydia K
Skagit Marriages 1884884 -- 19201930 Groom Last Name Groom First Name Bride Last Name Bride First Name Marriage Year Aadland Nels Paulson Lydia K. 1929 Aarstad Joakim Johnson Boe Anna 1922 Abbenhouse C. P.H. Rollofsen Rollofje 1911 Abbot Asa E. Gilden Gertrude A. 1906 Abbot Hollis R. Olson Esther C. 1907 Abbott John A. Ringdahl Prudence 1928 Abbott Linus Underwood Hariett L. 1891 Abbott Nelson S. Hoover Edith G. 1909 Abel Henry Houck Margaret 1920 Abel Joseph McNally Ellen 1893 Aberg Frans Alfred Petterson Augusta Elizabeth 1906 Abrams Milfred S. Hadden Gertrude 1915 Abrams Richard H. Anderson Mattie 1895 Achey Fred B. Dorcy Sara 1927 Adair James Whitehead Katherine 1910 Adam Valentine Bruns Margaret 1885 Adam Walter H. Bust Beatrice B. 1923 Adams E. O. Fredricks Reina 1903 Adams James P. Moody Pearl V. 1928 Adams Jimmy Louisa Mary 1894 Adams Jimmy Bob Betsy 1897 Adams Jimmy Peters Maggie 1911 Adams Regnial Shute Smith Maud S. 1903 Adams Val Trabue Naoma Oakes 1915 Adams William Burnerd Dunlap Minnie Myrtle 1909 Adamson Arvel T. Shoemaker Bessie Barbara 1926 Adcox William V. Koalmo Agnes 1925 Addington Marvin M. Lapham Ethel L. 1926 Adema Floyd M. Dowell Eva R. 1922 Adler William F. Fitch Geolak 1906 Admiral Pete Ruiter Johanna Wilhelmina 1927 Skagit Marriages 1884884 -- 19201930 Groom Last Name Groom First Name Bride Last Name Bride First Name Marriage Year Affleck Albert J. Affleck Effie Mae 1926 Ahn Edward S. Elde Tyra S. 1925 Ahrendt William G. Howell Ella 1927 Ahrens Marvin W. MacCarahan Eva 1925 Ainsworth Charles S. Hamrick Maude 1910 Ake Joseph Alexander Kimble Herma 1917 Akin Benjamin C. -
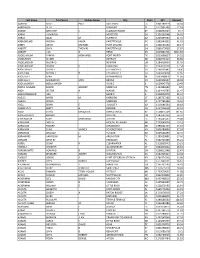
Last Name First Name Middle Name City State NPI Amount AARONS
Last Name First Name Middle Name City State NPI Amount AARONS SCOTT PAUL BAYTOWN TX 1841386919 14.64 AARONSON BETH DANBURY CT 1417991456 17.18 ABANG ANTHONY E ELIZABETHTOWN KY 1548250525 15.7 ABBAS HUMAYUN MODESTO CA 1336136548 14.01 ABBAS JALAL M SURPRISE AZ 1205895034 11.55 ABBASINEJAD MEISHA KAMA FAYETTEVILLE NC 1982646352 41.81 ABBEY DAVID MICHAEL FORT COLLINS CO 1568483196 26.17 ABBOTT JOHN THOMAS FAYETTEVILLE GA 1861679805 17.67 ABBOTT LISA G RENO NV 1609883701 6833.69 ABDEL HALIM AYMAN MOHAMED FORT WORTH TX 1093806697 14.23 ABDELHADY MAZEN DETROIT MI 1669771507 36.69 ABDELMESSIH MOURAD NEWARK OH 1184628950 51.33 ABDELMESSIH NIVEEN K GLENDALE CA 1922071059 17.54 ABDELSAYED GEORGE LOS ANGELES CA 1568721843 5600 ABDULIAN MICHAEL H LOS ANGELES CA 1649469628 12.66 ABDULLAH AZRA INDIANAPOLIS IN 1063468916 11.96 ABDULLAH GHAZANFAR SYED BRONX NY 1639108137 18.18 ABDULMASSIH ABDULMASSIH SKOKIE IL 1629045778 10.49 ABDUS-SALAAM SHARIF ASHANTI MEMPHIS TN 1134386592 125 ABDY VICTOR A WAYNE NJ 1164446795 11.47 ABEDMAHMOUD ISSA HERRIN IL 1528042975 25.34 ABELES ARYEH M. MERIDEN CT 1285737155 45.52 ABELES MICHA MERIDEN CT 1497751556 45.52 ABELL BRIAN E AUGUSTA GA 1215081534 18.43 ABERNATHY BRETT B. DENVER CO 1437106028 32.51 ABIDI SAIYID MANZOOR MAPLE SHADE NJ 1508816729 101.65 ABOUHOSSEIN AHMAD DAYTON OH 1487693156 56.07 ABOUMATAR SAMI MOHAMAD AUSTIN TX 1760445407 19.48 ABRAHAM BACHU MOUNT PLEASANT MI 1225045024 10.19 ABRAHAM ROBERT JONESBORO AR 1780641563 15.63 ABRAHAM SUNIL VARKEY SCHENECTADY NY 1801080890 37.03 ABRAHAM WILLIAM L TUCSON AZ -

5 Second Club
January February March April May June July August September October November December 1990 N/A N/A N/A Dave Phillips Dave Phillips Mike Koskan Ken Ashby Bob Blum N/A N/A William Jones Mark Thornton 1991 Ken Ashby Ken Ashby N/A Mike Koskan Kathy Erickson Mark Blaquiere John Ketchum John Ketchum Mike Koskan Ken Ashby Ken Ashby Ron Powers Robert Paddock John Ketchum Ron Powers Ron Powers James Gabhart Mark Blaquiere Glenn Noe Bill Turley Darwin Tangen Andy Beach 1992 John Ketchum Mike Koskan William Jones Mark Blaquiere Ken Ashby Ken Ashby Bill Turley James Gabhart W.D. Kopp Robert Paddock Jim Tressa Andy Beach Bill Turley Tim Blaquiere Jacquie Baldelli Ken Hamilton 1993 W.D Kopp Mike Koskan Mike Sullivan Debbie Tressa Mark Blaquiere Ken Ashby Debbie Tressa Debbie Mead Jim Tressa Ray Boydstun 1994 John Ketchum Laura Fossier John Ketchum Mike Koskan Ken Ashby Bob Patrick Kathy Erickson Mike Koskan John Ketchum Matt Ellis Whit Ellis Jim Tressa James Gabhart Ray Kirchmeyer Joan Phillips 1995 Rick Burbank Ken Ashby Jacquie Baldelli Mike Koskan Ken Ashby Mark Blaquiere Ray Kirchmeyer Bob Patrick John Ketchum Jacquie Baldelli Bob Wilmot Mike Burgie John Gibson James Gabhart Marylyn Patrick Richard Smith Richard Smith Jacquie Baldelli Richard Smith Chawki Belhadi Terry Ziegler Terry Mahlum Erin Koskan Terry Ziegler Marylyn Patrick Tom Munyan Ron Koskan Mike Jones Jon Johnson 1996 Ken Ashby John Ketchum Doug Adams Andy Beach Ken Ashby Mike Koskan Ken Ashby Mike Koskan Doug Adams Ken Ashby Joel Sampson Mike Koskan David Hall Terry Ziegler Tom Munyan -

Surname Given Name Book Name Page 32 Division Fat Memoirs 158, 159, 160 4-H Club River City Memoirs 5 90 4-H Clubs River City Memoirs 6 70 9Th Co
Surname Given Name Book Name Page 32 Division Fat Memoirs 158, 159, 160 4-H Club River City Memoirs 5 90 4-H Clubs River City Memoirs 6 70 9th Co. Replacement Training Troops Fat Memoirs 157 A & P Fat Memoirs 200 A & P River City Memoirs 6 88, 119 A & P River City Memoirs 2 65, 71 A & P River City Memoirs 3 86 A.F. of L. River City Memoirs 1 40 A.F.L. Unions River City Memoirs 6 12 Abbotsford, Wisconsin River City Memoirs 6 31, 106 Abbott Justin River City Memoirs 5 36 Abbott Sarah M. River City Memoirs 5 36 Abel Frank River City Memoirs 6 71 Abel Frank River City Memoirs 2 65 Abel's Clothes Store River City Memoirs 6 90, 119 Abel's Men Store River City Memoirs 6 88 Abitibi Fat Memoirs 183 Abolitionist Home Mission 25 Abrams Albert Fat Memoirs 118 Abrel Michal Home Mission 21 Ace of Club Flying Club River City Memoirs 6 65 Adams L. Earl Home Mission 113 Adams River City Memoirs 2 108 Adams County Fair grounds River City Memoirs 1 84 Adams County Historical Society River City Memoirs 1 84 Adams County School River City Memoirs 2 19 Adams County, Wisconsin Fat Memoirs 10, 11, 25, 26, 27, 110, 139, 158, 159, 196 Adams County, Wisconsin Home Mission 11, 30 Adams County, Wisconsin River City Memoirs 6 14, 25, 46, 104 Adams County, Wisconsin River City Memoirs 5 60, 82 Adams Express Company River City Memoirs 2 30 Adams Friendship River City Memoirs 1 84 Adams Mall River City Memoirs 2 108 Adams, Wisconsin Fat Memoirs 197 Adams, Wisconsin River City Memoirs 6 43 Adams-Friendship, Wisconsin River City Memoirs 6 16 Adams-Friendship, Wisconsin River City Memoirs 2 32 Adirondack Mountains Fat Memoirs 33 Adler Mildred River City Memoirs 6 65 Adler Beauty Shop Mildred Mrs. -

December JUNE GOOD NEWS
GOOD NEWS JUNE FIRST LUTHERAN DecemberFEBRUARYJUNE2017 CHURCH BOTTINEAU COUNTY FAIR 701 MAIN STREET June 15th—18th Food Safety Certification BOTTINEAU, ND June 4th at 10:15 a.m. 58318 (701) 228-2228 www.bottineauflc.org METIGOSHE DAY CAMP/VBS June 19th—23rd OUR MISSION STATEMENT Joyfully Growing as God’s People for Lives of Faith, Love, and Service To make arrangements to receive the newsletter, change your address, or to submit information for the newsletter or bulletin please call Janell at 228-2228 or e-mail her at Created by Janell Shannon Administrative Assistant 05-25-17 Page 2 A MESSAGE FROM OUR PASTOR Pastor Ben Carlsen Greetings, salutations, and hellos; let me introduce myself one more time – Pastor Ben Carlsen, called by all of you to serve as Pastor here at First Lutheran Church in Bottineau, ND. Some of you reading this have met me and introduced yourselves to me already, and to you I say “thank you” and “I’m sorry, but could you repeat your name.” And to those I have yet to meet, I look forward to being introduced and also asking repeatedly for your names. God may know each of us so intimately that the hairs of our heads are numbered, but for me, it’s going to take me a bit of time to learn your names and faces and the community connections, so I humbly ask for your patience and pardon should I neglect to put a face with a name or say “hello” on the street. Having said that, I am sure you are all aware of the effort to have a series of “meet and greet” events with medium size groups of congregational members.