Summary - Mitochondrial Myopathy Dr
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mitochondrial Trnaleu(Uur) May Cause an MERRF Syndrome
J7ournal ofNeurology, Neurosurgery, and Psychiatry 1996;61:47-51 47 The A to G transition at nt 3243 of the J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.61.1.47 on 1 July 1996. Downloaded from mitochondrial tRNALeu(uuR) may cause an MERRF syndrome Gian Maria Fabrizi, Elena Cardaioli, Gaetano Salvatore Grieco, Tiziana Cavallaro, Alessandro Malandrini, Letizia Manneschi, Maria Teresa Dotti, Antonio Federico, Giancarlo Guazzi Abstract Two distinct maternally inherited encephalo- Objective-To verify the phenotype to myopathies with ragged red fibres have been genotype correlations of mitochondrial recognised on clinical grounds: MERRF, DNA (mtDNA) related disorders in an which is characterised by myoclonic epilepsy, atypical maternally inherited encephalo- skeletal myopathy, neural deafness, and optic myopathy. atrophy,' and MELAS, which is defined by Methods-Neuroradiological, morpholog- stroke-like episodes in young age, episodic ical, biochemical, and molecular genetic headache and vomiting, seizures, dementia, analyses were performed on the affected lactic acidosis, skeletal myopathy, and short members of a pedigree harbouring the stature.2 Molecular genetic studies later con- heteroplasmic A to G transition at firmed the nosological distinction between the nucleotide 3243 of the mitochondrial two disorders, showing that MERRF is strictly tRNAI-u(UR), which is usually associated associated with two mutations of the mito- with the syndrome of mitochondrial chondrial tRNALYs at nucleotides 83443 and encephalomyopathy, lactic -

Cardiac Manifestations in Emery–Dreifuss Muscular Dystrophy
PRACTICE | CASES CPD Cardiac manifestations in Emery–Dreifuss muscular dystrophy Whitney Faiella MD, Ricardo Bessoudo MD n Cite as: CMAJ 2018 December 3;190:E1414-7. doi: 10.1503/cmaj.180410 35-year-old man with a known history of Emery–Dreifuss muscular dystrophy called emergency medical services KEY POINTS (EMS) while at work one morning, reporting palpitations, • Emery–Dreifuss muscular dystrophy is one of many lightheadedness,A fatigue and a rapid heart rate. On arrival by neuromuscular diseases with cardiac involvement, including EMS, his pulse was documented at 195–200 beats/min, and his bradyarrhythmia, tachyarrhythmia and cardiomyopathy, and rhythm strips showed ventricular tachycardia (Figure 1A). He involves an increased risk of sudden cardiac death. underwent cardioversion and was given a bolus of amiodarone, • A recently published scientific statement highlights key cardiac 150 mg intravenously. In the emergency department and during manifestations in various forms of neuromuscular diseases and includes detailed recommendations regarding screening, admission, his symptoms persisted with rhythm strips showing follow-up and treatment for each individual disease. recurrent episodes of sustained ventricular tachycardia (Fig- • Medical optimization of cardiac function and early detection of ure 1B). He was subsequently started on an amiodarone drip and arrhythmias with subsequent insertion of a pacemaker or oral metoprolol. Echocardiography performed during admission defibrillator can be life-saving in this patient population. showed dilated cardiomyopathy with severe systolic dysfunction and an estimated ejection fraction of 23%. Cardiac catheteriza- tion was performed to rule out an ischemic cause of the cardio- With respect to the patient’s diagnosis of Emery–Dreifuss mus- myopathy and showed normal coronary arteries. -

Multiple Presentation of Mitochondrial Disorders Arch Dis Child: First Published As 10.1136/Adc.81.3.209 on 1 September 1999
Arch Dis Child 1999;81:209–215 209 Multiple presentation of mitochondrial disorders Arch Dis Child: first published as 10.1136/adc.81.3.209 on 1 September 1999. Downloaded from Andreea Nissenkorn, Avraham Zeharia, Dorit Lev, Aviva Fatal-Valevski, Varda Barash, Alisa Gutman, Shaul Harel, Tally Lerman-Sagie Abstract The most severely aVected organs in mito- The aim of this study was to assess the chondrial disorders are those depending on heterogeneous clinical presentations of high rate aerobic metabolism—for example, children with mitochondrial disorders the brain, skeletal and cardiac muscle, the sen- evaluated at a metabolic neurogenetic sory organs, and the kidney.125 We aimed to clinic. The charts of 36 children with describe the great variety of symptomatology in highly suspected mitochondrial disorders patients with mitochondrial disorders in Israel, were reviewed. Thirty one children were and to compare this with the common clinical diagnosed as having a mitochondrial dis- presentations in other countries. order, based on a suggestive clinical pres- entation and at least one of the accepted Patients and methods laboratory criteria; however, in five chil- Thirty six consecutive patients (20 boys and 16 dren with no laboratory criteria the diag- girls) were evaluated at the paediatric neurol- nosis remained probable. All of the ogy clinic, Dana Children’s Hospital from patients had nervous system involvement. August 1994 to August 1996 and at the meta- Twenty seven patients also had dysfunc- bolic neurogenetic clinic, Wolfson Medical tion of other systems: sensory organs in 15 Center from September 1996 to June 1998 for patients, cardiovascular system in five, suspected mitochondrial disorders. -

Neuromuscular Disorders Neurology in Practice: Series Editors: Robert A
Neuromuscular Disorders neurology in practice: series editors: robert a. gross, department of neurology, university of rochester medical center, rochester, ny, usa jonathan w. mink, department of neurology, university of rochester medical center,rochester, ny, usa Neuromuscular Disorders edited by Rabi N. Tawil, MD Professor of Neurology University of Rochester Medical Center Rochester, NY, USA Shannon Venance, MD, PhD, FRCPCP Associate Professor of Neurology The University of Western Ontario London, Ontario, Canada A John Wiley & Sons, Ltd., Publication This edition fi rst published 2011, ® 2011 by Blackwell Publishing Ltd Blackwell Publishing was acquired by John Wiley & Sons in February 2007. Blackwell’s publishing program has been merged with Wiley’s global Scientifi c, Technical and Medical business to form Wiley-Blackwell. Registered offi ce: John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK Editorial offi ces: 9600 Garsington Road, Oxford, OX4 2DQ, UK The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK 111 River Street, Hoboken, NJ 07030-5774, USA For details of our global editorial offi ces, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com/wiley-blackwell The right of the author to be identifi ed as the author of this work has been asserted in accordance with the UK Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher. -

Adult-Onset Mitochondrial Myopathy J
Postgrad Med J: first published as 10.1136/pgmj.68.797.212 on 1 March 1992. Downloaded from Postgrad Med J (1992) 68, 212 - 215 © The Fellowship of Postgraduate Medicine, 1992 Adult-onset mitochondrial myopathy J. Fernandez-Sola, J. Casademont, J.M. Grau, F. Graus', F. Cardellach, E. Pedrol and A. Urbano-Marquez Muscle Research Group, Internal Medicine Service, and 'Department ofNeurology, Hospital Clinic, Villarroel 170, 08036 Barcelona, Spain Summary: Mitochondrial diseases are polymorphic entities which may affect many organs and systems. Skeletal muscle involvement is frequent in the context of systemic mitochondrial disease, but adult-onset pure mitochondrial myopathy appears to be rare. We report 3 patients with progressive skeletal mitochondrial myopathy starting in adult age. In all cases, the proximal myopathy was the only clinical feature. Mitochondrial pathology was confirmed by evidence of ragged-red fibres in muscle histochemistry, an abnormal mitochondrial morphology in electron microscopy and by exclusion of other underlying diseases. No deletions of mitochondrial DNA were found. We emphasize the need to look for a mitochondrial disorder in some non-specific myopathies starting in adult life. Introduction Mitochondrial diseases consist ofvarious polymor- These patients usually have proximal muscleby copyright. phic pathological entities which usually involve weakness and myalgia. Muscle atrophy of the many organs and systems. They represent a wide affected areas is frequent. The whole clinical pic- clinical, pathological and biochemical spectrum.' ture is not specific and there is a wide differential Involvement ofskeletal and ocular muscles, central diagnosis including inflammatory, toxic, neoplas- and peripheral nervous system, liver, heart, blood tic, metabolic and endocrine myopathies. -

MITOCHONDRIAL MYOPATHY Mitochondrial Myopathy
MITOCHONDRIAL MYOPATHY Mitochondrial Myopathy What are Symptoms of Mitochondrial What are Mitochondrial Myopathies? Myopathies? In general, there is nervous system impairment, eye problems, hearing problems, cardiac abnormalities, skeletal muscle abnormalities, and disorders of the Mitochondrial Myopathies are a group gastrointestinal tract. Some symptoms can be so of diseases that affect the mitochondria, mild that they’re hardly noticeable, while others are the tiny energy factories found inside life-threatening. Muscle weakness, muscle pain, almost all cells. These diseases interfere fatigue, lack of endurance, poor balance and dif- ficulty in swallowing may be experienced. with the function of muscles. Because mitochondria occur in all cells in the What is the age of onset? body, mitochondrial myopathies can The age varies according to disease. also interfere with the function of other What causes Mitochondrial Myopathies? organs in the body. The disease group A defect exists in either a mitochondrial gene or a is comprised of Kearns-Sayre syndrome gene in the cell nucleus that affects the functioning (KSS); Leigh’s syndrome; mitochondrial of the mitochondria. DNA depletion syndrome(MDS): Is this disease inherited? mitochondrial encephalomyopathy, In general, if the defect is in a mitochondrial gene, lactic acidosis and stroke-like episodes inheritance is from the mother only. If the defect is (MELAS); myoclonic epilepsy with in a nuclear gene, it may be inherited from either ragged red fibers (MERRF); mitochondrial the mother or father. Some of the mitochondrial myopathies are sporadic, meaning that the abnormal neurogastrointestinal encephalopathy gene only occurs in the affected person. It was not syndrome (MNGIE); neuropathy, ataxia inherited from a parent and will not be passed on to and retinitis pigmentosa (NARP); Pearson children. -

Blueprint Genetics Comprehensive Muscular Dystrophy / Myopathy Panel
Comprehensive Muscular Dystrophy / Myopathy Panel Test code: NE0701 Is a 125 gene panel that includes assessment of non-coding variants. In addition, it also includes the maternally inherited mitochondrial genome. Is ideal for patients with distal myopathy or a clinical suspicion of muscular dystrophy. Includes the smaller Nemaline Myopathy Panel, LGMD and Congenital Muscular Dystrophy Panel, Emery-Dreifuss Muscular Dystrophy Panel and Collagen Type VI-Related Disorders Panel. About Comprehensive Muscular Dystrophy / Myopathy Muscular dystrophies and myopathies are a complex group of neuromuscular or musculoskeletal disorders that typically result in progressive muscle weakness. The age of onset, affected muscle groups and additional symptoms depend on the type of the disease. Limb girdle muscular dystrophy (LGMD) is a group of disorders with atrophy and weakness of proximal limb girdle muscles, typically sparing the heart and bulbar muscles. However, cardiac and respiratory impairment may be observed in certain forms of LGMD. In congenital muscular dystrophy (CMD), the onset of muscle weakness typically presents in the newborn period or early infancy. Clinical severity, age of onset, and disease progression are highly variable among the different forms of LGMD/CMD. Phenotypes overlap both within CMD subtypes and among the congenital muscular dystrophies, congenital myopathies, and LGMDs. Emery-Dreifuss muscular dystrophy (EDMD) is a condition that affects mainly skeletal muscle and heart. Usually it presents in early childhood with contractures, which restrict the movement of certain joints – most often elbows, ankles, and neck. Most patients also experience slowly progressive muscle weakness and wasting, beginning with the upper arm and lower leg muscles and progressing to shoulders and hips. -

Diagnosis of Mitochondrial Diseases: Clinical and Histological Study of Sixty Patients with Ragged Red Fibers
Original Article Diagnosis of mitochondrial diseases: Clinical and histological study of sixty patients with ragged red fibers Sundaram Challa, Meena A. Kanikannan*, Murthy M. K. Jagarlapudi**, Venkateswar R. Bhoompally***, Mohandas Surath*** Departmetns of Pathology and *Neurology, Nizam’s Institute of Medical Sciences, Hyderabad. **Institute of Neurological Sciences, Care Hospital, ***L.V. Prasad Eye Institute, Hyderabad, India. Background: Mitochondrial diseases are caused by muta- group of patients. tions in mitochondrial or nuclear genes, or both and most Key Words: Mitochondrial disease, Ragged-red fiber, Pro- patients do not present with easily recognizable disorders. gressive external ophthalmoplegia, Kearns-Sayre syndrome, The characteristic morphologic change in muscle biopsy, Myoclonus epilepsy with ragged-red fibers, Heart block. ragged-red fibers (RRFs) provides an important clue to the diagnosis. Materials and Methods: Demographic data, pre- senting symptoms, neurological features, and investigative findings in 60 patients with ragged-red fibers (RRFs) on muscle biopsy, seen between January 1990 and December Introduction 2002, were analyzed. The authors applied the modified res- piratory chain (RC) diagnostic criteria retrospectively to de- Mitochondrial diseases with ragged-red muscle fibers (RRF) termine the number of cases fulfilling the diagnostic criteria as well as some without RRF, are caused by mutations in mi- of mitochondrial disease. Results: The most common clini- tochondrial or nuclear genes, or both, which are normally in- cal syndrome associated with RRFs on muscle biopsy was volved in the formation and maintenance of a functionally in- progressive external ophthalmoplegia (PEO) with or with- tact oxidative phosphorylation system in the mitochondrial out other signs, in 38 (63%) patients. Twenty-six patients inner membrane.1 These disorders present, with bewildering (43%) had only external ophthalmoplegia, 5 (8%) patients array of clinical presentations and are usually dominated by presented with encephalomyopathy. -

Exploration of the Mitochondria As a Potential Therapeutic Target in Duchenne Muscular Dystrophy
EXPLORATION OF THE MITOCHONDRIA AS A POTENTIAL THERAPEUTIC TARGET IN DUCHENNE MUSCULAR DYSTROPHY MEGHAN C HUGHES A DISSERTATION SUBMITTED TO THE FACULTY OF GRADUATE STUDIES IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY GRADUATE PROGRAM IN Kinesiology and Health Sciences YORK UNIVERSITY TORONTO, ONTARIO April 2019 Ó Meghan Hughes, 2019 Abstract Duchenne muscular dystrophy (DMD) is a progressive muscle wasting disease resulting from a mutation in the X-linked gene encoding the protein dystrophin. DMD is characterized by profound muscle weakness as degenerating muscle is replaced by fat and connective tissue. Early loss of ambulation followed by premature death due to cardiac and/or respiratory failure characterize the most debilitating aspects of DMD, a disease for which there is currently no cure. Limited success has been reported when treating DMD with gene based therapies. Current standard of care involves glucocorticoids, which target a secondary cellular myopathy; inflammation. While this line of treatment has provided promising benefits, these drugs present a variety of negative side effects for patients. As such, extensive research has been focused on identifying both therapeutic targets and corresponding novel therapies for the treatment of the DMD myopathy. The focus of this dissertation was to first determine the degree and precise mechanism of mitochondrial dysfunction in DMD followed by the evaluation of SBT-20, a mitochondrial- targeted peptide, as a therapeutic candidate for the treatment and prevention of DMD pathophysiology. In order to address these questions, we first comprehensively evaluated mitochondrial bioenergetics across a spectrum of oxidative and glycolytic muscles in the D2.B10- DMDmdx/2J mouse (D2.mdx) in early and late stages of disease progression. -

A Novel Mitochondrial DNA Deletion in a Chinese Girl with Kearns-Sayre
CASE A novel mitochondrial DNA deletion in a Chinese girl REPORT with Kearns-Sayre syndrome Eric KC Yau 丘健昌 KY Chan 陳國燕 Kearns-Sayre syndrome is a rare disorder often caused by mitochondrial DNA rearrangement. KM Au 區鑑明 The most commonly reported mitochondrial DNA deletion is 4977 bp in size spanning TC Chow 周達倉 nucleotides 8469 and 13447. The clinical signs of Kearns-Sayre syndrome include chronic YW Chan 陳恩和 progressive external ophthalmoplegia, retinitis pigmentosa, heart block and cerebellar ataxia, as well as other heterogeneous manifestations including neuromuscular problems and endocrine disorders. Cardiac conduction defects can develop insidiously, leading to sudden death sometimes if not promptly recognised. This report focuses on the diagnosis of Kearns- Sayre syndrome in a Chinese girl who presented initially with short stature, delayed puberty, insidious onset of ptosis and later with typical features of Kearns-Sayre syndrome including complete heart block. Genetic analysis disclosed a novel 7.2 kilobases deletion in muscle tissue. Mitochondrial diseases have heterogeneous phenotypes and mutational analysis has proven to be an effective tool for confirming the diagnosis. Introduction Kearns-Sayre syndrome (KSS) is a rare mitochondrial disorder with multisystem involvement affecting the eye, muscle, heart, endocrine, peripheral and central nervous systems. Typical clinical features include ptosis, ophthalmoplegia, pigmentary retinopathy, cardiac conduction defects and/or cardiomyopathy, sensorineural deafness, myopathy, ataxia, developmental delay or regression, and endocrine disorders.1 Most deletions have been 4977 bp in size.2 We report here a young Chinese girl with a clinical diagnosis of KSS associated with a novel large-scale mitochondrial DNA (mtDNA) deletion. Case report The patient was the only child of non-consanguineous Chinese parents. -

Deletion in Blood Mitochondria1 DNA in Kearns-Sayre Syndrome
003 1-399819213106-0557$03.00/0 PEDIATRIC RESEARCH Vol. 31, No. 6, 1992 Copyright O 1992 International Pediatric Research Foundation, Inc. Printed in (I.S. A. Deletion in Blood Mitochondria1 DNA in Kearns-Sayre Syndrome NATHAN FISCHEL-GHODSIAN, M. CHARLOTTE BOHLMAN. TONI R. PREZANT, JOHN M. GRAHAM, JR., STEPHEN D. CEDERBAUM, AND MATTHEW J. EDWARDS Ahmunsori Departrnmt of Pediatrics and Medical Genetics Birth Defect Center, Steven Spielberg Pediatric Reseurch Center, Cedars-Sinai Medical Center and UCLA School of'ikfedicine, Los Aizgeles, California 90048 [N.F.G., M.C.B., T.R.P., J.M.G.]; Department of Pedialrics and Psychiatry and the Mental Retardation Research Center, UCLA School vf Medicine, Lo.r .4nGpeles,California 90024 [S.P.C.];and Newcnstle and New South WTales Genetics Services, Waratah, New South Mfales, 24rtstralia/M.J.E.] ABSTRAa. Mitochondria1 DNA deletions have been Pearson's marrow-pancreas syndrome is phenotypically an described in the Kearns-Sayre syndrome (KSS) and the entirely different mitochondrial disorder, with refractory siderob- Pearson's marrow-pancreas syndrome. In some cases, the lastic anemia, exocrine pancreatic insufficiency, and liver dys- same 4 977-bp deletion has been identified in these two function, usually leading to death in infancy (8). However, the very different diseases. Therefore, it is not currently pos- molecular defects are similar to those seen in KSS, with the most sible to predict the clinical phenotype from the size or common 4 977-bp deletion from nt 8 482 to nt 13 460 being location of the deletion. Instead, differential tissue distri- identical in both disorders (8. -
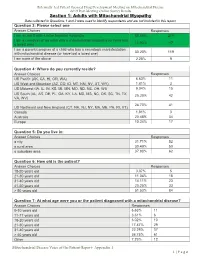
Section 1: Adults with Mitochondrial Myopathy Data Collected in Questions 1 and 2 Were Used to Identify Respondents and Are Not Included in This Report
Externally-Led Patient-Focused Drug Development Meeting on Mitochondrial Disease 2019 Post-Meeting Online Survey Results Section 1: Adults with Mitochondrial Myopathy Data collected in Questions 1 and 2 were used to identify respondents and are not included in this report. Question 3: Please select one Answer Choices Responses I am an adult with a mitochondrial myopathy 55.58% 219 I am a caregiver of an adult with a mitochondrial myopathy (or have lost a loved one) 11.93% 47 I am a parent/caregiver of a child who has a neurologic manisfestation with mitochondrial disease (or have lost a loved one) 30.20% 119 I am none of the above 2.28% 9 Question 4: Where do you currently reside? Answer Choices Responses US Pacific (AK, CA, HI, OR, WA) 6.63% 11 US West and Mountain (AZ, CO, ID, MT, NM, NV, UT, WY) 1.81% 3 US Midwest (IA, IL, IN, KS, MI, MN, MO, ND, NE, OH, WI) 9.04% 15 US South (AL, AR, DE, FL, GA, KY, LA, MD, MS, NC, OK, SC, TN, TX, 25.30% 42 VA, WV) 24.70% 41 US Northeast and New England (CT, NH, NJ, NY, MA, ME, PA, RI, VT) Canada 1.81% 3 Australia 20.48% 34 Europe 10.24% 17 Question 5: Do you live in: Answer Choices Responses a city 31.71% 52 a rural area 30.49% 50 a suburban area 37.80% 62 Question 6: How old is the patient? Answer Choices Responses 18-20 years old 3.07% 5 21-30 years old 11.04% 18 31-40 years old 14.11% 23 41-50 years old 20.25% 33 > 50 years old 51.53% 84 Question 7: At what age were you or the patient diagnosed with a mitochondrial disease? Answer Choices Responses 0-10 years old 6.63% 11 11-17 years old 3.61% 6 18-20 years old 6.02% 10 21-30 years old 17.47% 29 31-40 years old 22.29% 37 > 40 years old 36.75% 61 Other 7.23% 12 Mitochondrial Disease Voice of the Patient Report- Appendix 1 1 | P a g e Externally-Led Patient-Focused Drug Development Meeting on Mitochondrial Disease 2019 Post-Meeting Online Survey Results Question 8: Please select the answer that best describes the stage of physical disability for you or the person for whom you care Answer Choices Responses Minimal disability.