269721169-Oa
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of Intense Nest Predation in the Decline of Scarlet Robins and Eastern Yellow Robins in Remnant Woodland Near Armidale, New South Wales
The role of intense nest predation in the decline of Scarlet Robins and Eastern Yellow Robins in remnant woodland near Armidale, New South Wales S. J. S. DEBDSI A study of open-nesting Eastern Yellow Robins Eopsaltria australis and Scarlet Robins Petroica multicolor, on the New England Tablelands of New South Wales in 2000-02, found Iow breeding success typical of eucalypt woodland birds. The role of intense nest predation in the loss of birds from woodland fragments was investigated by means of predator-exclusion cages at robin nests, culling of Pied Currawongs Strepera graculina, and monitoring of fledging and recruitment in the robins. Nest-cages significantly improved nest success (86% vs 20%) and fledging rate (1.6 vs 0.3 fledglings per attempt) for both robin species combined (n = 7 caged, 20 uncaged). For both robin species combined, culling of currawongs produced a twofold difference in nest success (33% vs 14%), a higher fledging rate (0.5 vs 0.3 per attempt), and a five-day difference in mean nest survival (18 vs 13 days) (n = 62 nests), although sample sizes for nests in the cull treatment (n = 18) were small and nest predation continued. Although the robin breeding population had not increased one year after the cull, the pool of Yellow Robin recruits in 2001-03, after enhanced fledging success, produced two emigrants to a patch where Yellow Robins had become extinct. Management to assist the conservation of open-nesting woodland birds should address control of currawongs. Key words: Woodland birds, Habitat fragmentation, Nest predation, Predator exclusion, Predator removal. -

Disaggregation of Bird Families Listed on Cms Appendix Ii
Convention on the Conservation of Migratory Species of Wild Animals 2nd Meeting of the Sessional Committee of the CMS Scientific Council (ScC-SC2) Bonn, Germany, 10 – 14 July 2017 UNEP/CMS/ScC-SC2/Inf.3 DISAGGREGATION OF BIRD FAMILIES LISTED ON CMS APPENDIX II (Prepared by the Appointed Councillors for Birds) Summary: The first meeting of the Sessional Committee of the Scientific Council identified the adoption of a new standard reference for avian taxonomy as an opportunity to disaggregate the higher-level taxa listed on Appendix II and to identify those that are considered to be migratory species and that have an unfavourable conservation status. The current paper presents an initial analysis of the higher-level disaggregation using the Handbook of the Birds of the World/BirdLife International Illustrated Checklist of the Birds of the World Volumes 1 and 2 taxonomy, and identifies the challenges in completing the analysis to identify all of the migratory species and the corresponding Range States. The document has been prepared by the COP Appointed Scientific Councilors for Birds. This is a supplementary paper to COP document UNEP/CMS/COP12/Doc.25.3 on Taxonomy and Nomenclature UNEP/CMS/ScC-Sc2/Inf.3 DISAGGREGATION OF BIRD FAMILIES LISTED ON CMS APPENDIX II 1. Through Resolution 11.19, the Conference of Parties adopted as the standard reference for bird taxonomy and nomenclature for Non-Passerine species the Handbook of the Birds of the World/BirdLife International Illustrated Checklist of the Birds of the World, Volume 1: Non-Passerines, by Josep del Hoyo and Nigel J. Collar (2014); 2. -

Toxic Birds Not of a Feather
Commentary Avian chemical defense: Toxic birds not of a feather Paul J. Weldon Conservation and Research Center, Smithsonian Institution, 1500 Remount Road, Front Royal, VA 22630 n 1992, Dumbacher et al. (1) substan- Itially altered prevailing views of avian physiology, biochemistry, and chemical ecology with their report of the potent neurotoxin homobatrachotoxinin in feathers and other tissues of several spe- cies of New Guinean passerine birds of the genus Pitohui. Their discovery was signif- icant not only for suggesting a protective mechanism rarely considered for birds (i.e., chemical defense) but for the nature of the compound they discovered, a struc- turally complex alkaloid that binds Naϩ channels and depolarizes electrogenic membranes. Alkaloids in tetrapods gen- erally had been thought to be confined to amphibians, whose skins have long been acknowledged as arsenals of these biolog- Fig. 1. Hornets (Vespa orientalis) attacking a freshly skinned carcass of a laughing dove (Steptopelia ically active compounds (2). Indeed, be- senegalensis)(Left) while ignoring that of a pied kingfisher (Ceryle rudis). This observation prompted H. B. fore its discovery in Pitohui, homobatra- Cott (4) to undertake an extensive investigation of avian chemical defense. [Reproduced with permission chotoxinin, a member of a family of from ref. 4 (Copyright 1947, The Zoological Society of London).] steroidal alkaloids called batrachotoxinins (BTXs), had been found only in skin se- cretions of Central and South American dichrous), the most toxic of the birds they An additional enigma described by poison-dart frogs (Dendrobatidae) of the examined, is aposematic and may be Dumbacher et al. (3) is the profound genus Phyllobates. -

Nest, Egg, Incubation Behaviour and Vocalisations of the New Guinea Endemic Black Pitohui Melanorectes Nigrescens
Australian Field Ornithology 2019, 36, 116–120 http://dx.doi.org/10.20938/afo36116120 Nest, egg, incubation behaviour and vocalisations of the New Guinea endemic Black Pitohui Melanorectes nigrescens Richard H. Donaghey1, 2*, Donna J. Belder3 and Tony Baylis4 1Environmental Futures Research Institute, Griffith University, Nathan QLD 4111, Australia 280 Sawards Road, Myalla TAS 7325, Australia 3Fenner School of Environment and Society, The Australian National University, Canberra ACT 2601, Australia 4628 Utopia Road, Brooweena QLD 4621, Australia *Corresponding author. Email: [email protected] Abstract. The Black Pitohui Melanorectes nigrescens (Pachycephalidae) is endemic to mid-mountain forests in mainland New Guinea. Spectrograms of three different songs recorded in the Yopno Urawa Som Conservation Area (YUS CA), Huon Peninsula, Papua New Guinea, are presented. We describe the elevation and nest-site, height above the ground, structure and materials of a nest with an egg, discovered in the YUS CA. The colour and dimensions of the egg are documented. We present photographs of the nest-site, nest, and egg, and compare these with previously described eggs and a nest. We observed only the female Black Pitohui incubating the single-egg clutch. We compare the meagre information on pitohui breeding biology with that of Australian whistlers and shrike-thrushes. Introduction (Bell 1983). In lowland rainforest sites, flock composition was more influenced by the presence of Papuan Babblers Garritornis isidori (Pomatostomidae) than by that of The genus Melanorectes was resurrected by Dumbacher pitohuis, indicating that pitohui toxicity does not drive flock (2014) for the Black Pitohui, previously named Pitohui organisation (Goodale et al. 2012). -

Southwest Pacific Islands: Samoa, Fiji, Vanuatu & New Caledonia Trip Report 11Th to 31St July 2015
Southwest Pacific Islands: Samoa, Fiji, Vanuatu & New Caledonia Trip Report 11th to 31st July 2015 Orange Fruit Dove by K. David Bishop Trip Report - RBT Southwest Pacific Islands 2015 2 Tour Leaders: K. David Bishop and David Hoddinott Trip Report compiled by Tour Leader: K. David Bishop Tour Summary Rockjumper’s inaugural tour of the islands of the Southwest Pacific kicked off in style with dinner at the Stamford Airport Hotel in Sydney, Australia. The following morning we were soon winging our way north and eastwards to the ancient Gondwanaland of New Caledonia. Upon arrival we then drove south along a road more reminiscent of Europe, passing through lush farmlands seemingly devoid of indigenous birds. Happily this was soon rectified; after settling into our Noumea hotel and a delicious luncheon, we set off to explore a small nature reserve established around an important patch of scrub and mangroves. Here we quickly cottoned on to our first endemic, the rather underwhelming Grey-eared Honeyeater, together with Nankeen Night Herons, a migrant Sacred Kingfisher, White-bellied Woodswallow, Fantailed Gerygone and the resident form of Rufous Whistler. As we were to discover throughout this tour, in areas of less than pristine habitat we encountered several Grey-eared Honeyeater by David Hoddinott introduced species including Common Waxbill. And so began a series of early starts which were to typify this tour, though today everyone was up with added alacrity as we were heading to the globally important Rivierre Bleu Reserve and the haunt of the incomparable Kagu. We drove 1.3 hours to the reserve, passing through a stark landscape before arriving at the appointed time to meet my friend Jean-Marc, the reserve’s ornithologist and senior ranger. -

Gymnorhina Tibicen Global Invasive
FULL ACCOUNT FOR: Gymnorhina tibicen Gymnorhina tibicen System: Terrestrial Kingdom Phylum Class Order Family Animalia Chordata Aves Passeriformes Cracticidae Common name Synonym Coracias tibicen Similar species Grallina cyanoleuca, Cracticus nigrogularis Summary Gymnorhina tibicen (the Australian magpie) was originally introduced to New Zealand around the 1860s in an attempt to combat pastoral pests. It is known to be extremely territorial, especially during the breeding season, and is known to assault other avian species and even humans. Magpies potentially threaten a number of indigenous avian fauna, as well as putting humans at risk of injury. view this species on IUCN Red List Species Description The Australian magpie (Gymnorhina tibicen), is a medium-sized ground-feeding passerine found throughout much of the Australian continent. They range from 36-44cm in length and weigh 280-340g with black and white plumage, black heads, wings and underparts together with white napes and shoulders (NRC, 1998). The iris of the adult magpie is red, whereas the juveniles' eyes are dark brown in colour. Minor differences exist between the male and female magpies, though in general, magpies are not considered to be sexually dimorphic (Simpson et al., 1993). Notes Although Australian magpies (Gymnorhina tibicen) have been known to have detrimental impacts on some birds, they can actually be beneficial to others. They do this by attacking and displacing common avian predators, such as harrier hawks and ravens, which in turn provides safe nesting grounds for a number of rural birds (Morgan et al, 2005). Lifecycle Stages The average life span of the Australian magpie (Gymnorhina tibicen) has not been studied in detail, but is estimated to be around 24 years, with some individuals living up to 30 years of age (Reilly, 1988). -

New Guinean Passerines Have Globally Small Clutch-Sizes
Emu 2014 © BirdLife Australia 2014 doi:10.1071/MU14023_AC Supplementary material: Emu, 2014, 114(4), 295–303 New Guinean passerines have globally small clutch-sizes Benjamin G. FreemanA,B,C and Nicholas A. MasonA,B ADepartment of Ecology and Evolutionary Biology, Cornell University, Corson Hall, Ithaca, NY 14850, USA. BCornell Laboratory of Ornithology, 159 Sapsucker Woods Road, Ithaca, NY 14850, USA. CCorresponding author. Email: [email protected] Page 1 of 7 Emu 2014 © BirdLife Australia 2014 doi:10.1071/MU14023_AC Table S1. Clutch-size data for 102 species of passerines that occur in New Guinea Data from the Handbook of the Birds of the World: Alström et al. (2006), Boles (2006, 2007a, 2007b, 2007c), Cheke and Mann (2008), Coates (2008), Coates et al. (2006), Collar (2005), Frith and Frith (2009a, 2009b), Gregory (2007, 2008), Higgins et al. (2008), Rowley and Russell (2007, 2009), Taylor (2005), Van Balen (2008), Walther and Jones (2008) Scientific name Family Clutch-size Ailuroedus buccoides Ptilinorhynchidae 1 Ailuroedus melanotis Ptilonorhynchidae 1.5 Amblyornis macgregoriae Ptilonorhynchidae 1 Amblyornis inornata Ptilonorhynchidae 1 Archboldia papuensis Ptilonorhynchidae 1 Sericulus aureus Ptilonorhynchidae 1 Sipodotus wallacii Maluridae 2 Chenorhamphus grayi Maluridae 2 Xanthotis flaviventer Meliphagidae 2 Lichenostomus obscurus Meliphagidae 2 Meliphaga mimikae Meliphagidae 2 Meliphaga orientalis Meliphagidae 2 Meliphaga albonotata Meliphagidae 1.5 Meliphaga cinereifrons Meliphagidae 2 Meliphaga aruensis Meliphagidae 1.5 Philemon -
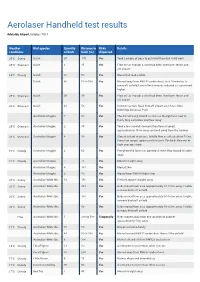
Aerolaser Handheld Test Results
Aerolaser Handheld test results Adelaide Airport October 2014 Weather Bird species Quantity Distance to Birds Details conditions of birds birds (m) dispersed 20°C Sunny Galah 20 170 Yes Took a couple of goes to get rid of them but it did work 20°C Overcast Galah 4 35 Yes Flew off as though a shot had been fired over fence and off airport 14°C Cloudy Galah 30 60 Yes Moved but took a while Galah 30 50 to 500 Yes Moved away from RWY12 undershoot, took 10 minutes to move off airfield, laser effectivenesds reduced as sun moved higher 20°C Overcast Galah 20 30 Yes Flew off as though a shot had been fired over fence and off airport 20°C Overcast Galah 40 50 Yes Instant reaction, flock flew off airport over fence intro Burbridge Business Park Australian Magpie 7 50 Yes The did not hang around as soon as the light was next to them, they carried on and flew away 20°C Overcast Australian Magpie 2 30 Yes Took a few seconds to react then flew at speed approximately 150m away and well away from the runway 20°C Overcast Australian Magpie 8 30 Yes Almost instant reactions. Initally flew as a flock about 100m. From that range I again used the laser. The birds flew out of sight over ops store 15°C Cloudy Australian Magpie 4 60 Yes Everytime the laser was pointed at them they moved straight away 15°C Cloudy Australian Magpie 7 70 Yes Moved straight away Australian Magpie 8 100 Yes Moved 24m Australian Magpie 2 50 Yes Moved from RWY05 flight strip 20°C Sunny Australian White Ibis 13 150 Yes Fled off airport straight away 20°C Sunny Australian White Ibis 250 Yes Birds moved from area approximately 50-100m away. -

New Caledonia, Fiji & Vanuatu
Field Guides Tour Report Part I: New Caledonia Sep 5, 2011 to Sep 15, 2011 Phil Gregory The revamped tour was a little later this year and it seemed to make some things a bit easier, note how well we did with the rare Crow Honeyeater, and Kagu was as ever a standout. One first-year bird was rewarded with a nice juicy scorpion that our guide found, and this really is a fabulous bird to see, another down on Harlan's famiy quest, too, as an added bonus to what is a quite unique bird. Cloven-feathered Dove was also truly memorable, and watching one give that strange, constipated hooting call was fantastic and this really is one of the world's best pigeons. Air Calin did their best to make life hard with a somewhat late flight to Lifou, and I have to say the contrast with the Aussie pilots in Vanuatu was remarkable -- these French guys must still be learning as they landed the ATR 42's so hard and had to brake so fiercely! Still, it all worked out and the day trip for the Ouvea Parakeet worked nicely, whilst the 2 endemic white-eyes on Lifou were got really early for once. Nice food, an interesting Kanak culture, with a trip to the amazing Renzo Piano-designed Tjibaou Cultural Center also feasible this The fantastic Kagu, star of the tour! (Photo by guide Phil year, and a relaxed pace make this a fun birding tour with some Gregory) terrific endemic birds as a bonus. My thanks to Karen at the Field Guides office for hard work on the complex logistics for this South Pacific tour, to the very helpful Armstrong at Arc en Ciel, Jean-Marc at Riviere Bleue, and to Harlan and Bart for helping me with my bags when I had a back problem. -

Diversification of Petroica Robins Across the Australo-Pacific Region: First Insights Into the Phylogenetic Affinities of New Guinea’S Highland Robin Species
Emu - Austral Ornithology ISSN: 0158-4197 (Print) 1448-5540 (Online) Journal homepage: http://www.tandfonline.com/loi/temu20 Diversification of Petroica robins across the Australo-Pacific region: first insights into the phylogenetic affinities of New Guinea’s highland robin species Anna M. Kearns, Leo Joseph, Aude Thierry, John F. Malloy, Maria Nandadevi Cortes-Rodriguez & Kevin E. Omland To cite this article: Anna M. Kearns, Leo Joseph, Aude Thierry, John F. Malloy, Maria Nandadevi Cortes-Rodriguez & Kevin E. Omland (2018): Diversification of Petroica robins across the Australo- Pacific region: first insights into the phylogenetic affinities of New Guinea’s highland robin species, Emu - Austral Ornithology, DOI: 10.1080/01584197.2018.1498744 To link to this article: https://doi.org/10.1080/01584197.2018.1498744 View supplementary material Published online: 06 Aug 2018. Submit your article to this journal View Crossmark data Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=temu20 EMU - AUSTRAL ORNITHOLOGY https://doi.org/10.1080/01584197.2018.1498744 Diversification of Petroica robins across the Australo-Pacific region: first insights into the phylogenetic affinities of New Guinea’s highland robin species Anna M. Kearns a,b, Leo Josephc, Aude Thierryd, John F. Malloya, Maria Nandadevi Cortes-Rodrigueza,e and Kevin E. Omlanda aDepartment of Biological Sciences, University of Maryland, Baltimore, USA; bCenter for Conservation Genomics, Smithsonian Conservation Biology Institute, National Zoological Park, Washington, DC, USA; cAustralian National Wildlife Collection, CSIRO National Research Collections Australia, Canberra, Australia; dSchool of Biological Sciences, University of Canterbury, Christchurch, New Zealand; eDepartment of Biology, Center for Natural Sciences, Ithaca College, Ithaca, USA ABSTRACT ARTICLE HISTORY Complex spatial and temporal phylogenetic patterns have emerged among Pacific Island radiations Received 13 December 2017 and their Australian and New Guinean congeners. -

The Currawongs and the Magpies
Diru’wunan and Diru’wun The Currawongs and the Magpies www.dharawalstories.com Frances Bodkin Gawaian Bodkin-Andrews Illustrations By Lorraine Robertson THE CURRAWONGS AND THE MAGPIES Diru’wunan and Diru’wun A very, very long time ago, Warnan’nan, the People of the Raven became worried. 1 There had not been any rain from the time of the blooming of the Boo’kerrikin to the time of the blooming of the Ker’wan. The creeks and rivers were drying up, and even some of the deepest waterholes were become shallow enough for children to walk across. And at night, the Earth sang a song of thirst, pleading for water to soothe her parched skin. Oftentimes, the People of the Raven would look at the skies, watching and hoping that the Cloud Spirits would come and deliver their bur- den of rain. But the skies remained blue and cloudless. The People of the Raven went to Wiritjiribin, the People of the Lyrebird, to seek their advice, but found that they, too were suffering from lack of rain, although their sweet water came from the Earth herself, and not from the skies like the other peoples. The People of the Lyrebird were kind to the People of the Raven and invited them to take as much sweet water as they needed. The People of the Raven asked why the People of the Lyrebird were so kind when other Peoples had refused to help them. The People of the Lyrebird replied that a long, long time ago, the People of the Raven had provided their black feathered cloaks to the children of the Mull’goh, the Black Swan, and providing the sweet water was the repayment of a kin debt. -

Eastern Australia: October-November 2016
Tropical Birding Trip Report Eastern Australia: October-November 2016 A Tropical Birding SET DEPARTURE tour EASTERN AUSTRALIA: From Top to Bottom 23rd October – 11th November 2016 The bird of the trip, the very impressive POWERFUL OWL Tour Leader: Laurie Ross All photos in this report were taken by Laurie Ross/Tropical Birding. 1 www.tropicalbirding.com +1-409-515-9110 [email protected] Page Tropical Birding Trip Report Eastern Australia: October-November 2016 INTRODUCTION The Eastern Australia Set Departure Tour introduces a huge amount of new birds and families to the majority of the group. We started the tour in Cairns in Far North Queensland, where we found ourselves surrounded by multiple habitats from the tidal mudflats of the Cairns Esplanade, the Great Barrier Reef and its sandy cays, lush lowland and highland rainforests of the Atherton Tablelands, and we even made it to the edge of the Outback near Mount Carbine; the next leg of the tour took us south to Southeast Queensland where we spent time in temperate rainforests and wet sclerophyll forests within Lamington National Park. The third, and my favorite leg, of the tour took us down to New South Wales, where we birded a huge variety of new habitats from coastal heathland to rocky shorelines and temperate rainforests in Royal National Park, to the mallee and brigalow of Inland New South Wales. The fourth and final leg of the tour saw us on the beautiful island state of Tasmania, where we found all 13 “Tassie” endemics. We had a huge list of highlights, from finding a roosting Lesser Sooty Owl in Malanda; to finding two roosting Powerful Owls near Brisbane; to having an Albert’s Lyrebird walk out in front of us at O Reilly’s; to seeing the rare and endangered Regent Honeyeaters in the Capertee Valley, and finding the endangered Swift Parrot on Bruny Island, in Tasmania.