Recent Advances in Engineering Nonribosomal Peptide Assembly Lines Cite This: Nat
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Review of Oxepine-Pyrimidinone-Ketopiperazine Type Nonribosomal Peptides
H OH metabolites OH Review Review of Oxepine-Pyrimidinone-Ketopiperazine Type Nonribosomal Peptides Yaojie Guo , Jens C. Frisvad and Thomas O. Larsen * Department of Biotechnology and Biomedicine, Technical University of Denmark, Søltofts Plads, Building 221, DK-2800 Kgs. Lyngby, Denmark; [email protected] (Y.G.); [email protected] (J.C.F.) * Correspondence: [email protected]; Tel.: +45-4525-2632 Received: 12 May 2020; Accepted: 8 June 2020; Published: 15 June 2020 Abstract: Recently, a rare class of nonribosomal peptides (NRPs) bearing a unique Oxepine-Pyrimidinone-Ketopiperazine (OPK) scaffold has been exclusively isolated from fungal sources. Based on the number of rings and conjugation systems on the backbone, it can be further categorized into three types A, B, and C. These compounds have been applied to various bioassays, and some have exhibited promising bioactivities like antifungal activity against phytopathogenic fungi and transcriptional activation on liver X receptor α. This review summarizes all the research related to natural OPK NRPs, including their biological sources, chemical structures, bioassays, as well as proposed biosynthetic mechanisms from 1988 to March 2020. The taxonomy of the fungal sources and chirality-related issues of these products are also discussed. Keywords: oxepine; nonribosomal peptides; bioactivity; biosynthesis; fungi; Aspergillus 1. Introduction Nonribosomal peptides (NRPs), mostly found in bacteria and fungi, are a class of peptidyl secondary metabolites biosynthesized by large modularly organized multienzyme complexes named nonribosomal peptide synthetases (NRPSs) [1]. These products are amongst the most structurally diverse secondary metabolites in nature; they exhibit a broad range of activities, which have been exploited in treatments such as the immunosuppressant cyclosporine A and the antibiotic daptomycin [2,3]. -

Amide Bond Formation in Nonribosomal Peptide Synthesis
Amide Bond Formation in Nonribosomal Peptide Synthesis: The Formylation and Condensation Domains Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) dem Fachbereich Chemie der Philipps-Universität Marburg vorgelegt von Georg Schönafinger aus München Marburg/Lahn 2007 Vom Fachbereich Chemie der Philipps-Universität Marburg als Dissertation am _______________ angenommen. Erstgutachter : Prof. Dr. M. A. Marahiel (Philipps-Universität, Marburg) Zweitgutachter : Prof. Dr. L.-O. Essen (Philipps-Universität, Marburg) Tag der Disputation: 20. Dezember 2007 2 The majority of the work presented here has been published: Samel, S.A.†, Schoenafinger, G. †, Knappe, T.A., Marahiel, M.A., Essen, L.O. 2007. Structural and functional insights into a peptide bond-forming bidomain from a nonribosomal peptide synthetase. Structure 15(7): 781-92. † equal contribution Schoenafinger, G., Schracke, N., Linne, U., Marahiel, M.A. 2006. Formylation domain: an essential modifying enzyme for the nonribosomal biosynthesis of linear gramicidin. J Am Chem Soc. 128(23): 7406-7. Schoenafinger, G., Marahiel, M.A. accepted 2007. Nonribosomal Peptides. Wiley Encyclopaedia of Chemical Biology. In press. 3 To Anke 4 Summary Nonribosomal peptides are of outstanding pharmacological interest, since many representatives of this highly diverse class of natural products exhibit therapeutically important activities, such as antibacterial, antitumor and immunosuppressive properties. Understanding their biosynthesis performed by multimodular mega-enzymes, the nonribosomal peptide synthetases (NRPSs), is one of the key determinants in order to be able to reprogram these machineries for the production of novel therapeutics. The central structural motif of all peptides is the peptide (or amide) bond. In this work, two different amide bond forming catalytic entities from NRPSs were studied: The condensation (C) and formylation (F) domains. -

Construction of Hybrid Peptide Synthetases for the Production of A-L-Aspartyl-L-Phenylalanine, a Precursor for the High-Intensity Sweetener Aspartame
Eur. J. Biochem. 270, 4555–4563 (2003) Ó FEBS 2003 doi:10.1046/j.1432-1033.2003.03858.x Construction of hybrid peptide synthetases for the production of a-L-aspartyl-L-phenylalanine, a precursor for the high-intensity sweetener aspartame Thomas Duerfahrt1, Sascha Doekel1,*, Theo Sonke2, Peter J. L. M. Quaedflieg2 and Mohamed A. Marahiel1 1Philipps-Universita¨t Marburg, Fachbereich Chemie/Biochemie, Marburg, Germany; 2DSM Research, Life Sciences – Advanced Synthesis, Catalysis and Development, Geleen, the Netherlands Microorganisms produce a large number of pharmaco- Product release was ensured by a C-terminally fused logically and biotechnologically important peptides by thioesterase domains and quantified by HPLC/MS ana- using nonribosomal peptide synthetases (NRPSs). Due to lysis. Significant differences of enzyme activity caused by their modular arrangement and their domain organization the fusion strategies were observed. Two forms of the NRPSs are particularly suitable for engineering recom- Asp-Phe dipeptide were detected, the expected a-Asp-Phe binant proteins for the production of novel peptides with and the by-product b-Asp-Phe. Dependent on the turn- interesting properties. In order to compare different over rates ranging from 0.01–0.7 min)1, the amount of strategies of domain assembling and module fusions we a-Asp-Phe was between 75 and 100% of overall product, focused on the selective construction of a set of peptide indicating a direct correlation between the turnover synthetases that catalyze the formation of the dipeptide numbers and the ratios of a-Asp-Phe to b-Asp-Phe. a-L-aspartyl-L-phenylalanine (Asp-Phe), the precursor of Taken together these results provide useful guidelines for the high-intensity sweetener a-L-aspartyl-L-phenylalanine the rational construction of hybrid peptide synthetases. -

Nonribosomal Peptides Synthetases and Their Applications in Industry Mario Alberto Martínez‑Núñez1,2* and Víctor Eric López Y López3
Martínez‑Núñez and López Sustain Chem Process (2016) 4:13 DOI 10.1186/s40508-016-0057-6 REVIEW Open Access Nonribosomal peptides synthetases and their applications in industry Mario Alberto Martínez‑Núñez1,2* and Víctor Eric López y López3 Abstract Nonribosomal peptides are products that fall into the class of secondary metabolites with a diverse properties as toxins, siderophores, pigments, or antibiotics, among others. Unlike other proteins, its biosynthesis is independent of ribosomal machinery. Nonribosomal peptides are synthesized on large nonribosomal peptide synthetase (NRPS) enzyme complexes. NRPSs are defined as multimodular enzymes, consisting of repeated modules. The NRPS enzymes are at operons and their regulation can be positive or negative at transcriptional or post-translational level. The pres‑ ence of NRPS enzymes has been reported in the three domains of life, being prevalent in bacteria. Nonribosomal pep‑ tides are use in human medicine, crop protection, or environment restoration; and their use as commercial products has been approved by the U. S Food and Drug Administration (FDA) and the U. S. Environmental Protection Agency (EPA). The key features of nonribosomal peptides and NRPS enzymes, and some of their applications in industry are summarized. Keywords: Nonribosomal peptides, Nonribosomal peptides synthetases, Environmental restoration, Human healt Background half of the NRPS enzymes found in a genome-mining Nonribosomal peptides is a diverse family of natural study of 2699 genomes by Wang et al. [1] are nonmodular products fall into the class of secondary metabolites with NRPS enzymes. Nonmodular NRPS enzymes are found a diverse properties as toxins, siderophores, pigments, in siderophore biosynthetic pathways like EntE and antibiotics, cytostatics, immunosuppressants or antican- VibH in enterobactin, and VibE in vibriobactin [5] or as cer agents [1, 2]; and have a particularity: their synthesis a stand-alone peptidyl carrier protein such as BlmI from is independent of ribosomal machinery. -

Biosynthesis and Engineering of Cyclomarin and Cyclomarazine: Prenylated, Non-Ribosomal Cyclic Peptides of Marine Actinobacterial Origin
UC San Diego Research Theses and Dissertations Title Biosynthesis and Engineering of Cyclomarin and Cyclomarazine: Prenylated, Non-Ribosomal Cyclic Peptides of Marine Actinobacterial Origin Permalink https://escholarship.org/uc/item/21b965z8 Author Schultz, Andrew W. Publication Date 2010 Peer reviewed eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Biosynthesis and Engineering of Cyclomarin and Cyclomarazine: Prenylated, Non-Ribosomal Cyclic Peptides of Marine Actinobacterial Origin A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Oceanography by Andrew William Schultz Committee in charge: Professor Bradley Moore, Chair Professor Eric Allen Professor Pieter Dorrestein Professor William Fenical Professor William Gerwick 2010 Copyright Andrew William Schultz, 2010 All rights reserved. The Dissertation of Andrew William Schultz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: ________________________________________________________________ ________________________________________________________________ ________________________________________________________________ Chair University of California, San Diego 2010 iii DEDICATION To my wife Elizabeth and our son Orion and To my parents Dale and Mary Thank you for your never ending love and support iv TABLE OF CONTENTS Signature Page .................................................................................................... -

Atlas of Nonribosomal Peptide and Polyketide Biosynthetic Pathways Reveals Common Occurrence of Nonmodular Enzymes
Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes Hao Wanga,1, David P. Fewera, Liisa Holmb, Leo Rouhiainena, and Kaarina Sivonena aDivision of Microbiology and Biotechnology, Department of Food and Environmental Sciences and bInstitute of Biotechnology and Department of Biosciences, Viikki Biocenter, University of Helsinki, FIN-00014, Helsinki, Finland Edited by Susan S. Golden, University of California, San Diego, La Jolla, CA, and approved May 12, 2014 (received for review January 27, 2014) Nonribosomal peptides and polyketides are a diverse group of adenylation, and acyl carrier domains for brucebactin biosynthesis natural products with complex chemical structures and enormous in Brucella abortus strain 2308 are encoded as fully separated pharmaceutical potential. They are synthesized on modular non- proteins (11); thus, these NRPSs could be considered to have ribosomal peptide synthetase (NRPS) and polyketide synthase type II architecture. The arrangement of modules within the (PKS) enzyme complexes by a conserved thiotemplate mechanism. NRPS and type I PKS enzymes often determines the number and Here, we report the widespread occurrence of NRPS and PKS genetic order of the monomer constituents of the product (12), despite machinery across the three domains of life with the discovery of deviations in module iteration (13, 14) and skipping (15). A 3,339 gene clusters from 991 organisms, by examining a total of growing number of gene clusters encoding both NRPSs and type I 2,699 genomes. These gene clusters display extraordinarily diverse PKSs have been identified for biosynthesis of complex natural organizations, and a total of 1,147 hybrid NRPS/PKS clusters were products (16). -
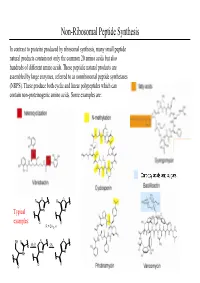
Non-Ribosomal Peptide Synthesis
Non-Ribosomal Peptide Synthesis In contrast to proteins produced by ribosomal synthesis, many small peptide natural products contain not only the common 20 amino acids but also hundreds of different amino acids. These peptidic natural products are assembled by large enzymes, referred to as nonribosomal peptide synthetases (NRPS). These produce both cyclic and linear polypeptides which can contain non-proteinogenic amino acids. Some examples are: O R R O Typical N N examples: O O R = CH3, H SH O S S -H2OOx. NH N N O O O Non-Ribosomal Peptide Synthesis: Initiation (A) Non-ribosomal peptide bio-synthesis requires A proper assembly and modification of a large multienzyme complexes including the addition of the prosthetic moiety 4'-phosphopantetheinyl cofactor (ppan) to a conserved serine residue of chain of peptidyl carrier protein (PCP). (B) The first step of peptide assembly is similar to ribosomal protein synthesis: an amino acid is B activated by trans-esterification with ATP to afford the corresponding aminoacyl-adenylate. The domains A1 and A2 each bind to a specific amino acid through non-covalent interactions and catalyze the trans-esterification to produce each aminoacyl- adenylate. “A” = adenylation domain. (C) Domain A also catalyzes the transfer of each C aminoacyl-adenylate onto the free thiol group of PCP- ppan within its module. This establishes a covalent linkage between enzyme and substrate. At this stage the substrate can undergo modifications such as epimerization or N-methylation by neighboring editing or “E” domains. Non-Ribosomal Peptide Synthesis: Elongation (D) Assembly of the product occurs by a series of D peptide bond formation steps (elongation) between the downstream building block with its free amine and the carboxy-thioester of the upstream substrate. -

Diversity of Linear Non-Ribosomal Peptide in Biocontrol Fungi
Journal of Fungi Review Diversity of Linear Non-Ribosomal Peptide in Biocontrol Fungi Xiaoyan Niu 1, Narit Thaochan 2 and Qiongbo Hu 1,* 1 Key Laboratory of Bio-Pesticide Innovation and Application of Guangdong Province, College of Agriculture, South China Agricultural University, Guangzhou 510642, China; [email protected] 2 Pest Management Biotechnology and Plant Physiology Laboratory, Faculty of Natural Resources, Prince of Songkla University, Hat Yai, Songkhla 90110, Thailand; [email protected] * Correspondence: [email protected] Received: 13 March 2020; Accepted: 9 May 2020; Published: 12 May 2020 Abstract: Biocontrol fungi (BFs) play a key role in regulation of pest populations. BFs produce multiple non-ribosomal peptides (NRPs) and other secondary metabolites that interact with pests, plants and microorganisms. NRPs—including linear and cyclic peptides (L-NRPs and C-NRPs)—are small peptides frequently containing special amino acids and other organic acids. They are biosynthesized in fungi through non-ribosomal peptide synthases (NRPSs). Compared with C-NRPs, L-NRPs have simpler structures, with only a linear chain and biosynthesis without cyclization. BFs mainly include entomopathogenic and mycoparasitic fungi, that are used to control insect pests and phytopathogens in fields, respectively. NRPs play an important role of in the interactions of BFs with insects or phytopathogens. On the other hand, the residues of NRPs may contaminate food through BFs activities in the environment. In recent decades, C-NRPs in BFs have been thoroughly reviewed. However, L-NRPs are rarely investigated. In order to better understand the species and potential problems of L-NRPs in BFs, this review lists the L-NRPs from entomopathogenic and mycoparasitic fungi, summarizes their sources, structures, activities and biosynthesis, and details risks and utilization prospects. -

The Specificity-Conferring Code of Adenylation Domains in Nonribosomal Peptide Synthetases Torsten Stachelhaus*, Henning D Mootz and Mohamed a Marahiel
Research Paper 493 The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases Torsten Stachelhaus*, Henning D Mootz and Mohamed A Marahiel Background: Many pharmacologically important peptides are synthesized Address: Biochemie/Fachbereich Chemie, nonribosomally by multimodular peptide synthetases (NRPSs). These enzyme Philipps-Universität Marburg, Hans-Meerwein- Straße, D-35032 Marburg, Germany. templates consist of iterated modules that, in their number and organization, determine the primary structure of the corresponding peptide products. At the *Present address: Department of Biological core of each module is an adenylation domain that recognizes the cognate Chemistry and Molecular Pharmacology, Harvard substrate and activates it as its aminoacyl adenylate. Recently, the crystal Medical School, 240 Longwood Avenue, Boston, structure of the phenylalanine-activating adenylation domain PheA was solved with MA 02115, USA. phenylalanine and AMP, illustrating the structural basis for substrate recognition. Correspondence: Mohamed A Marahiel E-mail: [email protected] Results: By comparing the residues that line the phenylalanine-binding pocket in PheA with the corresponding moieties in other adenylation domains, general rules Key words: adenylation domain, binding pocket, nonribosomal peptide synthetase, signature for deducing substrate specificity were developed. We tested these in silico sequence, substrate specificity ‘rules’ by mutating specificity-conferring residues within PheA. The substrate specificity of most mutants was altered or relaxed. Generalization of the selectivity Received: 11 March 1999 determinants also allowed the targeted specificity switch of an aspartate- Revisions requested: 13 April 1999 Revisions received: 27 April 1999 activating adenylation domain, the crystal structure of which has not yet been Accepted: 4 May 1999 solved, by introducing a single mutation. -

NIH Public Access Author Manuscript Proteins
NIH Public Access Author Manuscript Proteins. Author manuscript; available in PMC 2014 July 24. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Proteins. 2014 July ; 82(7): 1210–1218. The crystal structure of BlmI as a model for nonribosomal peptide synthetase peptidyl carrier proteins Jeremy R. Lohman1, Ming Ma1, Marianne E. Cuff2, Lance Bigelow2, Jessica Bearden2, Gyorgy Babnigg2, Andrzej Joachimiak2, George N. Phillips Jr.3, and Ben Shen1,4,5,* 1Department of Chemistry, The Scripps Research Institute, Jupiter, Florida 33458 2Midwest Center for Structural Genomics and Structural Biology Center, Biosciences Division, Argonne National Laboratory, Argonne, Illinois 60439 3Department of Biochemistry and Cell Biology, Rice University, Houston, Texas 77251 4Department of Molecular Therapeutics, The Scripps Research Institute, Jupiter, Florida 33458 5Natural Products Library Initiative at The Scripps Research Institute, The Scripps Research Institute, Jupiter, Florida 33458 Abstract Carrier proteins (CPs) play a critical role in the biosynthesis of various natural products, especially in nonribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) enzymology, where the CPs are referred to as peptidyl-carrier proteins (PCPs) or acyl-carrier proteins (ACPs), respectively. CPs can either be a domain in large multifunctional polypeptides or standalone proteins, termed Type I and Type II, respectively. There have been many biochemical studies of the Type I PKS and NRPS CPs, and of Type II ACPs. However, recently a number of Type II PCPs have been found and biochemically characterized. In order to understand the possible interaction surfaces for combinatorial biosynthetic efforts we crystallized the first characterized and representative Type II PCP member, BlmI, from the bleomycin biosynthetic pathway from Streptomyces verticillus ATCC 15003. -

Drosophila Melanogaster Nonribosomal Peptide Synthetase Ebony Encodes an Atypical Condensation Domain
Drosophila melanogaster nonribosomal peptide synthetase Ebony encodes an atypical condensation domain Thierry Izoréa,b,c,1, Julien Tailhadesa,b,c, Mathias Henning Hansena,b,c, Joe A. Kaczmarskid, Colin J. Jacksond, and Max J. Crylea,b,c,1 aThe Monash Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia; bDepartment of Biochemistry and Molecular Biology, Monash University, Clayton, VIC 3800, Australia; cEMBL Australia, Monash University, Clayton, VIC 3800, Australia; and dResearch School of Chemistry, The Australian National University, Acton, ACT 2601, Australia Edited by Mohamed A. Marahiel, Philipps-Universität Marburg, Marburg, Germany, and accepted by Editorial Board Member Michael A. Marletta December 27, 2018 (received for review July 5, 2018) The protein Ebony from Drosophila melanogaster plays a central peptide and the PCP-bound amino acid. This then leads to the role in the regulation of histamine and dopamine in various tissues transfer of the peptide from the upstream PCP to the down- through condensation of these amines with β-alanine. Ebony is a stream PCP and extends the peptide by one residue (3). In ad- rare example of a nonribosomal peptide synthetase (NRPS) from a dition to these essential domains, most NRPSs encode a terminal higher eukaryote and contains a C-terminal sequence that does thioesterase domain (TE domain) in the last module of the as- not correspond to any previously characterized NRPS domain. sembly line to allow the release of the product from the synthesis We have structurally characterized this C-terminal domain and machinery. The TE domain also serves as a point for further di- have discovered that it adopts the aryl-alkylamine-N-acetyl trans- versification of the peptide sequence through various pathways, ferase (AANAT) fold, which is unprecedented in NRPS biology. -

Distribution and Evolution of Nonribosomal Peptide Synthetase Gene Clusters in the Ceratocystidaceae
G C A T T A C G G C A T genes Article Distribution and Evolution of Nonribosomal Peptide Synthetase Gene Clusters in the Ceratocystidaceae Mohammad Sayari , Magriet A. van der Nest, Emma T. Steenkamp, Nicole C. Soal, P. Markus Wilken and Brenda D. Wingfield * Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute, University of Pretoria, Pretoria 0002, South Africa; [email protected] (M.S.); [email protected] (M.A.v.d.N.); [email protected] (E.T.S.); [email protected] (N.C.S.); [email protected] (P.M.W.) * Correspondence: brenda.wingfi[email protected]; Tel.: +27-0124206288 Received: 6 March 2019; Accepted: 1 April 2019; Published: 30 April 2019 Abstract: In filamentous fungi, genes in secondary metabolite biosynthetic pathways are generally clustered. In the case of those pathways involved in nonribosomal peptide production, a nonribosomal peptide synthetase (NRPS) gene is commonly found as a main element of the cluster. Large multifunctional enzymes are encoded by members of this gene family that produce a broad spectrum of bioactive compounds. In this research, we applied genome-based identification of nonribosomal peptide biosynthetic gene clusters in the family Ceratocystidaceae. For this purpose, we used the whole genome sequences of species from the genera Ceratocystis, Davidsoniella, Thielaviopsis, Endoconidiophora, Bretziella, Huntiella, and Ambrosiella. To identify and characterize the clusters, different bioinformatics and phylogenetic approaches, as well as PCR-based methods were used. In all genomes studied, two highly conserved NRPS genes (one monomodular and one multimodular) were identified and their potential products were predicted to be siderophores.