Supplementary Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Studies on the Proteome of Human Hair - Identifcation of Histones and Deamidated Keratins Received: 15 August 2017 Sunil S
www.nature.com/scientificreports OPEN Studies on the Proteome of Human Hair - Identifcation of Histones and Deamidated Keratins Received: 15 August 2017 Sunil S. Adav 1, Roopa S. Subbaiaih2, Swat Kim Kerk 2, Amelia Yilin Lee 2,3, Hui Ying Lai3,4, Accepted: 12 January 2018 Kee Woei Ng3,4,7, Siu Kwan Sze 1 & Artur Schmidtchen2,5,6 Published: xx xx xxxx Human hair is laminar-fbrous tissue and an evolutionarily old keratinization product of follicle trichocytes. Studies on the hair proteome can give new insights into hair function and lead to the development of novel biomarkers for hair in health and disease. Human hair proteins were extracted by detergent and detergent-free techniques. We adopted a shotgun proteomics approach, which demonstrated a large extractability and variety of hair proteins after detergent extraction. We found an enrichment of keratin, keratin-associated proteins (KAPs), and intermediate flament proteins, which were part of protein networks associated with response to stress, innate immunity, epidermis development, and the hair cycle. Our analysis also revealed a signifcant deamidation of keratin type I and II, and KAPs. The hair shafts were found to contain several types of histones, which are well known to exert antimicrobial activity. Analysis of the hair proteome, particularly its composition, protein abundances, deamidated hair proteins, and modifcation sites, may ofer a novel approach to explore potential biomarkers of hair health quality, hair diseases, and aging. Hair is an important and evolutionarily conserved structure. It originates from hair follicles deep within the der- mis and is mainly composed of hair keratins and KAPs, which form a complex network that contributes to the rigidity and mechanical properties. -

Long Non-Coding RNA Landscape in Prostate Cancer Molecular Subtypes: a Feature Selection Approach
International Journal of Molecular Sciences Article Long Non-Coding RNA Landscape in Prostate Cancer Molecular Subtypes: A Feature Selection Approach Simona De Summa 1,* , Antonio Palazzo 2 , Mariapia Caputo 1, Rosa Maria Iacobazzi 3 , Brunella Pilato 1, Letizia Porcelli 3, Stefania Tommasi 1 , Angelo Virgilio Paradiso 4,† and Amalia Azzariti 3,† 1 Molecular Diagnostics and Pharmacogenetics Unit, IRCCS IstitutoTumori Giovanni Paolo II, 70124 Bari, Italy; [email protected] (M.C.); [email protected] (B.P.); [email protected] (S.T.) 2 Laboratory of Nanotechnology, IRCCS IstitutoTumori Giovanni Paolo II, 70124 Bari, Italy; [email protected] 3 Laboratory of Experimental Pharmacology, IRCCS Istituto Tumori Giovanni Paolo II, 70124 Bari, Italy; [email protected] (R.M.I.); [email protected] (L.P.); [email protected] (A.A.) 4 Scientific Directorate, IRCCS Istituto Tumori Giovanni Paolo II, 70124 Bari, Italy; [email protected] * Correspondence: [email protected] † Co-senior authors. Abstract: Prostate cancer is one of the most common malignancies in men. It is characterized by a high molecular genomic heterogeneity and, thus, molecular subtypes, that, to date, have not been used in clinical practice. In the present paper, we aimed to better stratify prostate cancer patients through the selection of robust long non-coding RNAs. To fulfill the purpose of the study, a bioinformatic approach focused on feature selection applied to a TCGA dataset was used. In such a way, LINC00668 and long non-coding(lnc)-SAYSD1-1, able to discriminate ERG/not-ERG subtypes, Citation: De Summa, S.; Palazzo, A.; were demonstrated to be positive prognostic biomarkers in ERG-positive patients. -

University of Alberta
University of Alberta T cell-mediated inflammation is stereotyped: mouse delayed-type hypersensitivity reaction and mouse T cell-mediated rejection of renal allografts share common molecular mechanisms by Jeffery M. Venner A thesis submitted to the Faculty of Graduate Studies and Research in partial fulfillment of the requirements for the degree of Master of Science in Experimental Medicine Department of Medicine ©Jeffery M. Venner Fall 2011 Edmonton, Alberta Permission is hereby granted to the University of Alberta Libraries to reproduce single copies of this thesis and to lend or sell such copies for private, scholarly or scientific research purposes only. Where the thesis is converted to, or otherwise made available in digital form, the University of Alberta will advise potential users of the thesis of these terms. The author reserves all other publication and other rights in association with the copyright in the thesis and, except as herein before provided, neither the thesis nor any substantial portion thereof may be printed or otherwise reproduced in any material form whatsoever without the author's prior written permission. Dedicated to those who have inspired me: My family and friends, my teachers and mentors. You laid the foundation – my success is guaranteed ABSTRACT Genome-wide gene expression analysis of diseases has revealed large-scale changes in the expression of thousands of genes (transcripts) representing biological processes. The processes that occur during T cell-mediated rejection (TCMR) of renal allografts in mice and humans have been previously delineated, and they appear to be independent of cytotoxic mechanisms; thus, TCMR is analogous to delayed-type hypersensitivity (DTH). -

A Genomics Approach Reveals Insights Into the Importance of Gene Losses for Mammalian Adaptations
Corrected: Publisher correction ARTICLE DOI: 10.1038/s41467-018-03667-1 OPEN A genomics approach reveals insights into the importance of gene losses for mammalian adaptations Virag Sharma1,2,3, Nikolai Hecker1,2,3, Juliana G. Roscito1,2,3, Leo Foerster1,2,3, Bjoern E. Langer1,2,3 & Michael Hiller1,2,3 1234567890():,; Identifying the genomic changes that underlie phenotypic adaptations is a key challenge in evolutionary biology and genomics. Loss of protein-coding genes is one type of genomic change with the potential to affect phenotypic evolution. Here, we develop a genomics approach to accurately detect gene losses and investigate their importance for adaptive evolution in mammals. We discover a number of gene losses that likely contributed to morphological, physiological, and metabolic adaptations in aquatic and flying mammals. These gene losses shed light on possible molecular and cellular mechanisms that underlie these adaptive phenotypes. In addition, we show that gene loss events that occur as a consequence of relaxed selection following adaptation provide novel insights into species’ biology. Our results suggest that gene loss is an evolutionary mechanism for adaptation that may be more widespread than previously anticipated. Hence, investigating gene losses has great potential to reveal the genomic basis underlying macroevolutionary changes. 1 Max Planck Institute of Molecular Cell Biology and Genetics, Pfotenhauerstr. 108, 01307 Dresden, Germany. 2 Max Planck Institute for the Physics of Complex Systems, Noethnitzer Str. 38, 01187 Dresden, Germany. 3 Center for Systems Biology Dresden, Pfotenhauerstr. 108, 01307 Dresden, Germany. Correspondence and requests for materials should be addressed to M.H. (email: [email protected]) NATURE COMMUNICATIONS | (2018) 9:1215 | DOI: 10.1038/s41467-018-03667-1 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03667-1 ne of the most fascinating aspects of nature is the a b % conserved genes diversity of life. -

Array Painting Reveals a High Frequency of Balanced Translocations in Breast Cancer Cell Lines That Break in Cancer-Relevant Genes
Oncogene (2008) 27, 3345–3359 & 2008 Nature Publishing Group All rights reserved 0950-9232/08 $30.00 www.nature.com/onc ONCOGENOMICS Array painting reveals a high frequency of balanced translocations in breast cancer cell lines that break in cancer-relevant genes KD Howarth1, KA Blood1,BLNg2, JC Beavis1, Y Chua1, SL Cooke1, S Raby1, K Ichimura3, VP Collins3, NP Carter2 and PAW Edwards1 1Department of Pathology, Hutchison-MRC Research Centre, University of Cambridge, Cambridge, UK; 2Wellcome Trust Sanger Institute, Cambridge, UK and 3Department of Pathology, Division of Molecular Histopathology, Addenbrookes Hospital, University of Cambridge, Cambridge, UK Chromosome translocations in the common epithelial tion and inversion, which can result in gene fusion, cancers are abundant, yet little is known about them. promoter insertion or gene inactivation. As is well They have been thought to be almost all unbalanced and known in haematopoietic tumours and sarcomas, therefore dismissed as mostly mediating tumour suppres- translocations and inversions can have powerful onco- sor loss. We present a comprehensive analysis by array genic effects on specific genes and play a central role in painting of the chromosome translocations of breast cancer development (Rowley, 1998). In the past there cancer cell lines HCC1806, HCC1187 and ZR-75-30. In has been an implicit assumption that such rearrange- array painting, chromosomes are isolated by flow ments are not significant players in the common cytometry, amplified and hybridized to DNA microarrays. epithelial -

Identification of Novel Wool Keratin Intermediate Filament Genes in Sheep Skin Z-D
222 Yu et al. – Wool keratin genes Identification of novel wool keratin intermediate filament genes in sheep skin Z-D. YU1*, S.W. GORDON1, 2, J.E. WILDERMOTH1, O.A.M. WALLACE1, A.J. NIXON1 and A.J. PEARSON1 1AgResearch Ruakura, Private Bag 3123, Hamilton 3240, New Zealand 2Excerpta Medica, Sing Pao Building New Wing, 101 King's Road, North Point, Hong Kong *Corresponding author: [email protected] ABSTRACT Keratin intermediate filament (KIF) genes encode key proteins for hair and wool formation. A total of 17 expressed human KIF genes involved in hair formation are annotated. However, to date, only eight wool KIF genes have been reported. In this study, six new cDNA sequences (KRT32, KRT33B, KRT34, KRT39, KRT40 and KRT82) representing previously unreported wool KIFs were identified using a contiguous ovine sequence library constructed primarily from ESTs. The expression of three other KIF genes (KRT36, KRT84 and KRT87) was confirmed by PCR using sheep skin total RNA. The analogue of human KRT37 (type I) was unable to be identified, while KRT87 (type II) was present in sheep but not in humans. Therefore 10 type I and seven type II KIF family members have been identified in sheep in comparison to 11 and six KIFs in the human. These 17 KIF genes are likely to represent the complete or near-complete set involved in wool formation. The annotation of these genes will facilitate investigation into their patterns of expression in wool follicles and their roles in the determination of fibre attributes. Key words: keratin intermediate filament; wool; gene expression. -

The Genetics of Hair Shaft Disorders
CONTINUING MEDICAL EDUCATION The genetics of hair shaft disorders AmyS.Cheng,MD,a and Susan J. Bayliss, MDb,c Saint Louis, Missouri Many of the genes causing hair shaft defects have recently been elucidated. This continuing medical education article discusses the major types of hair shaft defects and associated syndromes and includes a review of histologic features, diagnostic modalities, and findings in the field of genetics, biochemistry, and molecular biology. Although genetic hair shaft abnormalities are uncommon in general dermatology practice, new information about genetic causes has allowed for a better understanding of the underlying pathophysiologies. ( J Am Acad Dermatol 2008;59:1-22.) Learning objective: At the conclusion of this article, the reader should be familiar with the clinical presentation and histologic characteristics of hair shaft defects and associated genetic diseases. The reader should be able to recognize disorders with hair shaft abnormalities, conduct appropriate referrals and order appropriate tests in disease evaluation, and select the best treatment or supportive care for patients with hair shaft defects. EVALUATION OF THE HAIR progresses via interactions with the mesenchymal For the student of hair abnormalities, a full review dermal papillae, leading to the formation of anagen of microscopic findings and basic anatomy can be hairs with complete follicular components, including found in the textbook Disorders of Hair Growth by sebaceous and apocrine glands.3 Elise Olsen,1 especially the chapter on ‘‘Hair Shaft Anagen hair. The hair shaft is composed of three Disorders’’ by David Whiting, which offers a thor- layers, called the medulla, cortex, and cuticle (Fig 1). ough review of the subject.1 The recognition of the The medulla lies in the center of the shaft and anatomic characteristics of normal hair and the effects contains granules with citrulline, an amino acid, of environmental factors are important when evalu- which is unique to the medulla and internal root ating a patient for hair abnormalities. -

Exploring Molecular Mechanisms Controlling Skin Homeostasis and Hair Growth
Exploring Molecular Mechanisms Controlling Skin Homeostasis and Hair Growth. MicroRNAs in Hair-cycle-Dependent Gene Regulation, Hair Growth and Associated Tissue Remodelling. Item Type Thesis Authors Ahmed, Mohammed I. Rights <a rel="license" href="http://creativecommons.org/licenses/ by-nc-nd/3.0/"><img alt="Creative Commons License" style="border-width:0" src="http://i.creativecommons.org/l/by- nc-nd/3.0/88x31.png" /></a><br />The University of Bradford theses are licenced under a <a rel="license" href="http:// creativecommons.org/licenses/by-nc-nd/3.0/">Creative Commons Licence</a>. Download date 02/10/2021 08:52:54 Link to Item http://hdl.handle.net/10454/5204 University of Bradford eThesis This thesis is hosted in Bradford Scholars – The University of Bradford Open Access repository. Visit the repository for full metadata or to contact the repository team © University of Bradford. This work is licenced for reuse under a Creative Commons Licence. Exploring Molecular Mechanisms Controlling Skin Homeostasis and Hair Growth MicroRNAs in Hair-cycle-Dependent Gene Regulation, Hair Growth and Associated Tissue Remodelling Mohammed Ikram AHMED BSc, MSc Submitted for the degree of Doctor of Philosophy Centre for Skin Sciences Division of Biomedical Sciences School of life Sciences University of Bradford 2010 Dedicated To my daughter Misba Ahmed and my Family II Abud-Darda (May Allah be pleased with him) reported; The messenger of Allah (PBUH) said, “He who follows a path in quest of knowledge, Allah will make the path of Jannah (heaven) easy to him. The angels lower their wings over the seeker of knowledge, being pleased with what he does. -
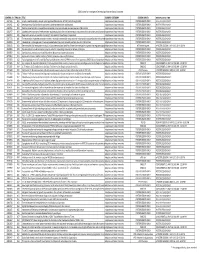
2020 ABSTRACT STATUS NOTIFICATION GRID.Xlsx
2020 Society for Investigative Dermatology Abstract Status Outcomes CONTROL ID FINAL ID TITLE CURRENT CATEGORY SESSION ORDER SESSION, DATE, TIME 3327155 001 Lymph node‐fibroblastic reticular cells regulate differentiation of CD4 T cells through CD25 Adaptive and Auto‐Immunity POSTER SESSION ONLY POSTER SESSION ONLY 3342451 002 Development of Guillain‐Barré syndrome in patients treated with adalimumab Adaptive and Auto‐Immunity POSTER SESSION ONLY POSTER SESSION ONLY 3352903 003 Dextran‐based acitretin nanoparticle ameliorates imiquimod‐induced psoriasis‐like skin inflammation Adaptive and Auto‐Immunity POSTER SESSION ONLY POSTER SESSION ONLY 3352977 004 Localized administration of methotrexate regulates psoriasis‐like skin inflammation and protects from secondary sensitization aAdaptive and Auto‐Immunity POSTER SESSION ONLY POSTER SESSION ONLY 3359994 005 Respiratory activity in psoriatic circulating T cells predicts the efficacy of apremilast Adaptive and Auto‐Immunity POSTER SESSION ONLY POSTER SESSION ONLY 3367733 006 The association of platelet activation markers, neutrophil extracellular traps and anti‐mitochondrial autoantibodies with cutaneAdaptive and Auto‐Immunity POSTER SESSION ONLY POSTER SESSION ONLY 3368003 007 Indoleamine 2,3‐dioxygenase 2 knockout exacerbates imiquimod‐induced psoriasis‐like skin inflammation Adaptive and Auto‐Immunity POSTER SESSION ONLY POSTER SESSION ONLY 3368100 008 Genome‐wide DNA methylation analysis in lupus keratinocytes identifies differential methylation of genes that regulate apoptoAdaptive and -

The Effects of Organic and Harsh Cleaners on Anolis Carolinensis
THE EFFECTS OF ORGANIC AND HARSH CLEANERS ON ANOLIS Formatted: Font:Italic CAROLINENSIS INTEGUMENT Formatted: Font:Italic A Report of a Senior Study by Kristen Rolston Major: Biology Maryville College Spring, 2017 Date approved , by Faculty Supervisor Date approved , by Division Chair ABSTRACT Household bleach has been used in the home for cleaning hard surfaces since the 18th century and has caused a number of injuries over time. This study further investigates how detrimental this caustic substance can be to the epidermis of Anolis carolinensis in Formatted: Font:Italic comparison to organically branded products claiming to be safer. Bleach cause significantly Deleted: It was determined that there was a significant effect reduced cell width (p=0.001) and number (p=0.009) on stratum corneum , whereas the Deleted: cell width (p=2.59x10-14) and number (p=0.009) organic cleaner showed no difference. This study illustrates that Anolis carolinensis are an Deleted: between the bleach and control groups appropriate model organism to examine the effects of certain substances on the Deleted: By understanding integumentary system and how the skin recovers could lead to valuable insight in the fields of dermatology and stem cell research. PAGE NUMBERS ARE INCORRECT THROUGHOUT 2 ACKNOWLEDGEMENTS ADD ACKNOWLEDGEMENTS HERE Deleted: [This section is not required. If included, it has a 2” top margin.] 3 TABLE OF CONTENTS Page List of Tables vi List of Figures vii Chapter I Introduction 1 Chapter II Title of Chapter 2 Chapter III Title of Chapter 3 Chapter IV Title of Chapter 4 Appendix (or Appendices, as appropriate) 5 Works Cited 7 FIX THIS Deleted: [This section has a 2” top margin.] 4 LIST OF TABLES Table Page 1. -

Characterization of a Novel Human Type II Epithelial Keratin K1b, Specifically Expressed in Eccrine Sweat Glands
Characterization of a Novel Human Type II Epithelial Keratin K1b, Specifically Expressed in Eccrine Sweat Glands Lutz Langbein,Ã Michael A. Rogers,w Silke Praetzel,Ã Bernard Cribier,z Bernard Peltre,z Nikolaus Gassler,y and Ju¨ rgen Schweizerw ÃDivision of Cell Biology and wSection of Normal and Neoplastic Epidermal Differentiation, German Cancer Research Center, Heidelberg, Germany; zDepartment of Dermatology, University of Strasbourg, Strasbourg, France; yInstitute of Pathology, University of Heidelberg, Heidelberg, Germany In this study, we show that a novel human type II epithelial keratin, K1b, is exclusively expressed in luminal duct cells of eccrine sweat glands. Taking this luminal K1b expression as a reference, we have used antibodies against a plethora of epithelial keratins to systematically investigate their expression in the secretory globule and the two- layered sweat duct, which was divided into the intraglandular, intradermal, and intraepidermal (acrosyringium) segments, the latter being further subdivided into the sweat duct ridge and upper intraepidermal duct. We show that (i) each of the eccrine sweat gland tissue compartments expresses their own keratin patterns, (ii) the peripheral and luminal duct layers exhibit a sequential keratin expression, with both representing self-renewing cell layers, (iii) the intradermal duct and the sweat duct ridge display hitherto unknown length variations, and (iv) out of all cell layers, the luminal cell layer is the most robust layer and expresses the highest number of keratins, these being concentrated at the apical side of the cells to form the cuticle. We provide evidence that the cellular and inter- cellular properties of the peripheral and the luminal layers reflect adaptations to different functions. -

Detection of H3k4me3 Identifies Neurohiv Signatures, Genomic
viruses Article Detection of H3K4me3 Identifies NeuroHIV Signatures, Genomic Effects of Methamphetamine and Addiction Pathways in Postmortem HIV+ Brain Specimens that Are Not Amenable to Transcriptome Analysis Liana Basova 1, Alexander Lindsey 1, Anne Marie McGovern 1, Ronald J. Ellis 2 and Maria Cecilia Garibaldi Marcondes 1,* 1 San Diego Biomedical Research Institute, San Diego, CA 92121, USA; [email protected] (L.B.); [email protected] (A.L.); [email protected] (A.M.M.) 2 Departments of Neurosciences and Psychiatry, University of California San Diego, San Diego, CA 92103, USA; [email protected] * Correspondence: [email protected] Abstract: Human postmortem specimens are extremely valuable resources for investigating trans- lational hypotheses. Tissue repositories collect clinically assessed specimens from people with and without HIV, including age, viral load, treatments, substance use patterns and cognitive functions. One challenge is the limited number of specimens suitable for transcriptional studies, mainly due to poor RNA quality resulting from long postmortem intervals. We hypothesized that epigenomic Citation: Basova, L.; Lindsey, A.; signatures would be more stable than RNA for assessing global changes associated with outcomes McGovern, A.M.; Ellis, R.J.; of interest. We found that H3K27Ac or RNA Polymerase (Pol) were not consistently detected by Marcondes, M.C.G. Detection of H3K4me3 Identifies NeuroHIV Chromatin Immunoprecipitation (ChIP), while the enhancer H3K4me3 histone modification was Signatures, Genomic Effects of abundant and stable up to the 72 h postmortem. We tested our ability to use H3K4me3 in human Methamphetamine and Addiction prefrontal cortex from HIV+ individuals meeting criteria for methamphetamine use disorder or not Pathways in Postmortem HIV+ Brain (Meth +/−) which exhibited poor RNA quality and were not suitable for transcriptional profiling.