Considerations for Feature Selection Using Gene Pairs and Applications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The N-Cadherin Interactome in Primary Cardiomyocytes As Defined Using Quantitative Proximity Proteomics Yang Li1,*, Chelsea D
© 2019. Published by The Company of Biologists Ltd | Journal of Cell Science (2019) 132, jcs221606. doi:10.1242/jcs.221606 TOOLS AND RESOURCES The N-cadherin interactome in primary cardiomyocytes as defined using quantitative proximity proteomics Yang Li1,*, Chelsea D. Merkel1,*, Xuemei Zeng2, Jonathon A. Heier1, Pamela S. Cantrell2, Mai Sun2, Donna B. Stolz1, Simon C. Watkins1, Nathan A. Yates1,2,3 and Adam V. Kwiatkowski1,‡ ABSTRACT requires multiple adhesion, cytoskeletal and signaling proteins, The junctional complexes that couple cardiomyocytes must transmit and mutations in these proteins can cause cardiomyopathies (Ehler, the mechanical forces of contraction while maintaining adhesive 2018). However, the molecular composition of ICD junctional homeostasis. The adherens junction (AJ) connects the actomyosin complexes remains poorly defined. – networks of neighboring cardiomyocytes and is required for proper The core of the AJ is the cadherin catenin complex (Halbleib and heart function. Yet little is known about the molecular composition of the Nelson, 2006; Ratheesh and Yap, 2012). Classical cadherins are cardiomyocyte AJ or how it is organized to function under mechanical single-pass transmembrane proteins with an extracellular domain that load. Here, we define the architecture, dynamics and proteome of mediates calcium-dependent homotypic interactions. The adhesive the cardiomyocyte AJ. Mouse neonatal cardiomyocytes assemble properties of classical cadherins are driven by the recruitment of stable AJs along intercellular contacts with organizational and cytosolic catenin proteins to the cadherin tail, with p120-catenin β structural hallmarks similar to mature contacts. We combine (CTNND1) binding to the juxta-membrane domain and -catenin β quantitative mass spectrometry with proximity labeling to identify the (CTNNB1) binding to the distal part of the tail. -

Systematic Data-Querying of Large Pediatric Biorepository Identifies Novel Ehlers-Danlos Syndrome Variant Akshatha Desai1, John J
Desai et al. BMC Musculoskeletal Disorders (2016) 17:80 DOI 10.1186/s12891-016-0936-8 RESEARCH ARTICLE Open Access Systematic data-querying of large pediatric biorepository identifies novel Ehlers-Danlos Syndrome variant Akshatha Desai1, John J. Connolly1, Michael March1, Cuiping Hou1, Rosetta Chiavacci1, Cecilia Kim1, Gholson Lyon1, Dexter Hadley1 and Hakon Hakonarson1,2* Abstract Background: Ehlers Danlos Syndrome is a rare form of inherited connective tissue disorder, which primarily affects skin, joints, muscle, and blood cells. The current study aimed at finding the mutation that causing EDS type VII C also known as “Dermatosparaxis” in this family. Methods: Through systematic data querying of the electronic medical records (EMRs) of over 80,000 individuals, we recently identified an EDS family that indicate an autosomal dominant inheritance. The family was consented for genomic analysis of their de-identified data. After a negative screen for known mutations, we performed whole genome sequencing on the male proband, his affected father, and unaffected mother. We filtered the list of non- synonymous variants that are common between the affected individuals. Results: The analysis of non-synonymous variants lead to identifying a novel mutation in the ADAMTSL2 (p. Gly421Ser) gene in the affected individuals. Sanger sequencing confirmed the mutation. Conclusion: Our work is significant not only because it sheds new light on the pathophysiology of EDS for the affected family and the field at large, but also because it demonstrates the utility of unbiased large-scale clinical recruitment in deciphering the genetic etiology of rare mendelian diseases. With unbiased large-scale clinical recruitment we strive to sequence as many rare mendelian diseases as possible, and this work in EDS serves as a successful proof of concept to that effect. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Targeting PH Domain Proteins for Cancer Therapy
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 12-2018 Targeting PH domain proteins for cancer therapy Zhi Tan Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Medicinal Chemistry and Pharmaceutics Commons, Neoplasms Commons, and the Pharmacology Commons Recommended Citation Tan, Zhi, "Targeting PH domain proteins for cancer therapy" (2018). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 910. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/910 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. TARGETING PH DOMAIN PROTEINS FOR CANCER THERAPY by Zhi Tan Approval page APPROVED: _____________________________________________ Advisory Professor, Shuxing Zhang, Ph.D. _____________________________________________ -

BRG1 Knockdown Inhibits Proliferation Through Multiple Cellular Pathways in Prostate Cancer Katherine A
Giles et al. Clin Epigenet (2021) 13:37 https://doi.org/10.1186/s13148-021-01023-7 RESEARCH Open Access BRG1 knockdown inhibits proliferation through multiple cellular pathways in prostate cancer Katherine A. Giles1,2,3, Cathryn M. Gould1, Joanna Achinger‑Kawecka1,4, Scott G. Page2, Georgia R. Kafer2, Samuel Rogers2, Phuc‑Loi Luu1,4, Anthony J. Cesare2, Susan J. Clark1,4† and Phillippa C. Taberlay3*† Abstract Background: BRG1 (encoded by SMARCA4) is a catalytic component of the SWI/SNF chromatin remodelling com‑ plex, with key roles in modulating DNA accessibility. Dysregulation of BRG1 is observed, but functionally uncharacter‑ ised, in a wide range of malignancies. We have probed the functions of BRG1 on a background of prostate cancer to investigate how BRG1 controls gene expression programmes and cancer cell behaviour. Results: Our investigation of SMARCA4 revealed that BRG1 is over‑expressed in the majority of the 486 tumours from The Cancer Genome Atlas prostate cohort, as well as in a complementary panel of 21 prostate cell lines. Next, we utilised a temporal model of BRG1 depletion to investigate the molecular efects on global transcription programmes. Depleting BRG1 had no impact on alternative splicing and conferred only modest efect on global expression. How‑ ever, of the transcriptional changes that occurred, most manifested as down‑regulated expression. Deeper examina‑ tion found the common thread linking down‑regulated genes was involvement in proliferation, including several known to increase prostate cancer proliferation (KLK2, PCAT1 and VAV3). Interestingly, the promoters of genes driving proliferation were bound by BRG1 as well as the transcription factors, AR and FOXA1. -

Genomic and Transcriptome Analysis Revealing an Oncogenic Functional Module in Meningiomas
Neurosurg Focus 35 (6):E3, 2013 ©AANS, 2013 Genomic and transcriptome analysis revealing an oncogenic functional module in meningiomas XIAO CHANG, PH.D.,1 LINGLING SHI, PH.D.,2 FAN GAO, PH.D.,1 JONATHAN RUssIN, M.D.,3 LIYUN ZENG, PH.D.,1 SHUHAN HE, B.S.,3 THOMAS C. CHEN, M.D.,3 STEVEN L. GIANNOTTA, M.D.,3 DANIEL J. WEISENBERGER, PH.D.,4 GAbrIEL ZADA, M.D.,3 KAI WANG, PH.D.,1,5,6 AND WIllIAM J. MAck, M.D.1,3 1Zilkha Neurogenetic Institute, Keck School of Medicine, University of Southern California, Los Angeles, California; 2GHM Institute of CNS Regeneration, Jinan University, Guangzhou, China; 3Department of Neurosurgery, Keck School of Medicine, University of Southern California, Los Angeles, California; 4USC Epigenome Center, Keck School of Medicine, University of Southern California, Los Angeles, California; 5Department of Psychiatry, Keck School of Medicine, University of Southern California, Los Angeles, California; and 6Division of Bioinformatics, Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, California Object. Meningiomas are among the most common primary adult brain tumors. Although typically benign, roughly 2%–5% display malignant pathological features. The key molecular pathways involved in malignant trans- formation remain to be determined. Methods. Illumina expression microarrays were used to assess gene expression levels, and Illumina single- nucleotide polymorphism arrays were used to identify copy number variants in benign, atypical, and malignant me- ningiomas (19 tumors, including 4 malignant ones). The authors also reanalyzed 2 expression data sets generated on Affymetrix microarrays (n = 68, including 6 malignant ones; n = 56, including 3 malignant ones). -
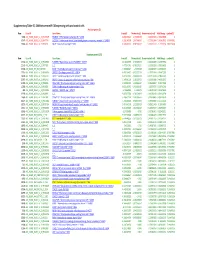
Suppl Table S5 SAM Between Mir138 and Control.Xlsx
Supplementary Table S5. SAM between miR‐138 expressing cells and control cells Positive genes (3) Row Gene ID Gene Name Score(d) Numerator(r) Denominator(s+s0) Fold Change q‐value(%) 5281 AGI_HUM1_OLIGO_A_23_P148175 FRMD4B ^ FERM domain containing 4B ^ 23150 8.891251624 1.476259259 0.166035033 2.782263885 0 35147 AGI_HUM1_OLIGO_A_32_P47754 SLC2A14 ^ Solute carrier family 2 (facilitated glucose transporter), member 3 ^ 144195 6.218260998 1.697166667 0.272932684 3.242635062 3.79547861 22089 AGI_HUM1_OLIGO_A_23_P76731 RAGE ^ Renal tumor antigen ^ 5891 4.905690362 0.785166667 0.160052227 1.72329139 7.85271438 Negative genes (121) Row Gene ID Gene Name Score(d) Numerator(r) Denominator(s+s0) Fold Change q‐value(%) 17132 AGI_HUM1_OLIGO_A_23_P408913 FLJ30990 ^ Hypothetical protein FLJ30990 ^ 150737 ‐13.42362905 ‐2.765333333 0.206004898 0.147079356 0 22142 AGI_HUM1_OLIGO_A_23_P77192 ^ ^ ‐7.473541761 ‐0.765237134 0.102392836 0.588356653 0 37434 AGI_OLIGO_NKI_NM_007036 ESM1 ^ Endothelial cell‐specific molecule 1 ^ 11082 ‐7.353800827 ‐2.9739038 0.404403637 0.127281637 0 15110 AGI_HUM1_OLIGO_A_23_P361014 ZNF537 ^ Zinc finger protein 537 ^ 57616 ‐6.453194027 ‐0.953237134 0.147715554 0.516472295 0 18428 AGI_HUM1_OLIGO_A_23_P44724 CSRP2 ^ Cysteine and glycine‐rich protein 2 ^ 1466 ‐6.421072015 ‐2.068333333 0.322116514 0.238434791 0 23767 AGI_HUM1_OLIGO_A_23_P92410 CASP3 ^ Caspase 3, apoptosis‐related cysteine protease ^ 836 ‐6.147096129 ‐1.183333333 0.192502819 0.440332937 0 21195 AGI_HUM1_OLIGO_A_23_P68486 C20orf108 ^ Chromosome 20 open reading frame -

Supplementary Table 1 Double Treatment Vs Single Treatment
Supplementary table 1 Double treatment vs single treatment Probe ID Symbol Gene name P value Fold change TC0500007292.hg.1 NIM1K NIM1 serine/threonine protein kinase 1.05E-04 5.02 HTA2-neg-47424007_st NA NA 3.44E-03 4.11 HTA2-pos-3475282_st NA NA 3.30E-03 3.24 TC0X00007013.hg.1 MPC1L mitochondrial pyruvate carrier 1-like 5.22E-03 3.21 TC0200010447.hg.1 CASP8 caspase 8, apoptosis-related cysteine peptidase 3.54E-03 2.46 TC0400008390.hg.1 LRIT3 leucine-rich repeat, immunoglobulin-like and transmembrane domains 3 1.86E-03 2.41 TC1700011905.hg.1 DNAH17 dynein, axonemal, heavy chain 17 1.81E-04 2.40 TC0600012064.hg.1 GCM1 glial cells missing homolog 1 (Drosophila) 2.81E-03 2.39 TC0100015789.hg.1 POGZ Transcript Identified by AceView, Entrez Gene ID(s) 23126 3.64E-04 2.38 TC1300010039.hg.1 NEK5 NIMA-related kinase 5 3.39E-03 2.36 TC0900008222.hg.1 STX17 syntaxin 17 1.08E-03 2.29 TC1700012355.hg.1 KRBA2 KRAB-A domain containing 2 5.98E-03 2.28 HTA2-neg-47424044_st NA NA 5.94E-03 2.24 HTA2-neg-47424360_st NA NA 2.12E-03 2.22 TC0800010802.hg.1 C8orf89 chromosome 8 open reading frame 89 6.51E-04 2.20 TC1500010745.hg.1 POLR2M polymerase (RNA) II (DNA directed) polypeptide M 5.19E-03 2.20 TC1500007409.hg.1 GCNT3 glucosaminyl (N-acetyl) transferase 3, mucin type 6.48E-03 2.17 TC2200007132.hg.1 RFPL3 ret finger protein-like 3 5.91E-05 2.17 HTA2-neg-47424024_st NA NA 2.45E-03 2.16 TC0200010474.hg.1 KIAA2012 KIAA2012 5.20E-03 2.16 TC1100007216.hg.1 PRRG4 proline rich Gla (G-carboxyglutamic acid) 4 (transmembrane) 7.43E-03 2.15 TC0400012977.hg.1 SH3D19 -

Integrated Bioinformatics Analysis Reveals Novel Key Biomarkers and Potential Candidate Small Molecule Drugs in Gestational Diabetes Mellitus
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.09.434569; this version posted March 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Integrated bioinformatics analysis reveals novel key biomarkers and potential candidate small molecule drugs in gestational diabetes mellitus Basavaraj Vastrad1, Chanabasayya Vastrad*2, Anandkumar Tengli3 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. 3. Department of Pharmaceutical Chemistry, JSS College of Pharmacy, Mysuru and JSS Academy of Higher Education & Research, Mysuru, Karnataka, 570015, India * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.03.09.434569; this version posted March 10, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Gestational diabetes mellitus (GDM) is one of the metabolic diseases during pregnancy. The identification of the central molecular mechanisms liable for the disease pathogenesis might lead to the advancement of new therapeutic options. The current investigation aimed to identify central differentially expressed genes (DEGs) in GDM. The transcription profiling by array data (E-MTAB-6418) was obtained from the ArrayExpress database. The DEGs between GDM samples and non GDM samples were analyzed with limma package. Gene ontology (GO) and REACTOME enrichment analysis were performed using ToppGene. Then we constructed the protein-protein interaction (PPI) network of DEGs by the Search Tool for the Retrieval of Interacting Genes database (STRING) and module analysis was performed. -
Gene Expression Profiling in Hepatocellular Carcinoma: Upregulation of Genes in Amplified Chromosome Regions
Modern Pathology (2008) 21, 505–516 & 2008 USCAP, Inc All rights reserved 0893-3952/08 $30.00 www.modernpathology.org Gene expression profiling in hepatocellular carcinoma: upregulation of genes in amplified chromosome regions Britta Skawran1, Doris Steinemann1, Anja Weigmann1, Peer Flemming2, Thomas Becker3, Jakobus Flik4, Hans Kreipe2, Brigitte Schlegelberger1 and Ludwig Wilkens1,2 1Institute of Cell and Molecular Pathology, Hannover Medical School, Hannover, Germany; 2Institute of Pathology, Hannover Medical School, Hannover, Germany; 3Department of Visceral and Transplantation Surgery, Hannover Medical School, Hannover, Germany and 4Institute of Virology, Hannover Medical School, Hannover, Germany Cytogenetics of hepatocellular carcinoma and adenoma have revealed gains of chromosome 1q as a significant differentiating factor. However, no studies are available comparing these amplification events with gene expression. Therefore, gene expression profiling was performed on tumours cytogenetically well characterized by array-based comparative genomic hybridisation. For this approach analysis was carried out on 24 hepatocellular carcinoma and 8 hepatocellular adenoma cytogenetically characterised by array-based comparative genomic hybridisation. Expression profiles of mRNA were determined using a genome-wide microarray containing 43 000 spots. Hierarchical clustering analysis branched all hepatocellular adenoma from hepatocellular carcinoma. Significance analysis of microarray demonstrated 722 dysregulated genes in hepatocellular carcinoma. Gene set enrichment analysis detected groups of upregulated genes located in chromosome bands 1q22–42 seen also as the most frequently gained regions by comparative genomic hybridisation. Comparison of significance analysis of microarray and gene set enrichment analysis narrowed down the number of dysregulated genes to 18, with 7 genes localised on 1q22 (SCAMP3, IQGAP3, PYGO2, GPATC4, ASH1L, APOA1BP, and CCT3). In hepatocellular adenoma 26 genes in bands 11p15, 11q12, and 12p13 were upregulated. -
SUPPLEMENTARY DATA Supplementary Table 1. Characteristics of the Organ Donors and Human Islet Preparations Used for RNA-Seq
SUPPLEMENTARY DATA Supplementary Table 1. Characteristics of the organ donors and human islet preparations used for RNA-seq and independent confirmation and mechanistic studies. Gender Age BMI Cause of death Purity (years) (kg/m2) (%) F 77 23.8 Trauma 45 M 36 26.3 CVD 51 M 77 25.2 CVD 62 F 46 22.5 CVD 60 M 40 26.2 Trauma 34 M 59 26.7 NA 58 M 51 26.2 Trauma 54 F 79 29.7 CH 21 M 68 27.5 CH 42 F 76 25.4 CH 30 F 75 29.4 CVD 24 F 73 30.0 CVD 16 M 63 NA NA 46 F 64 23.4 CH 76 M 69 25.1 CH 68 F 23 19.7 Trauma 70 M 47 27.7 CVD 48 F 65 24.6 CH 58 F 87 21.5 Trauma 61 F 72 23.9 CH 62 M 69 25 CVD 85 M 85 25.5 CH 39 M 59 27.7 Trauma 56 F 76 19.5 CH 35 F 50 20.2 CH 70 F 42 23 CVD 48 M 52 24.5 CH 60 F 79 27.5 CH 89 M 56 24.7 Cerebral ischemia 47 M 69 24.2 CVD 57 F 79 28.1 Trauma 61 M 79 23.7 NA 13 M 82 23 CH 61 M 32 NA NA 75 F 23 22.5 Cardiac arrest 46 M 51 NA Trauma 37 Abbreviations: F: Female; M: Male; BMI: Body mass index; CVD: Cardiovascular disease; CH: Cerebral hemorrhage. -

Proteomic Landscape of the Human Choroid–Retinal Pigment Epithelial Complex
Supplementary Online Content Skeie JM, Mahajan VB. Proteomic landscape of the human choroid–retinal pigment epithelial complex. JAMA Ophthalmol. Published online July 24, 2014. doi:10.1001/jamaophthalmol.2014.2065. eFigure 1. Choroid–retinal pigment epithelial (RPE) proteomic analysis pipeline. eFigure 2. Gene ontology (GO) distributions and pathway analysis of human choroid– retinal pigment epithelial (RPE) protein show tissue similarity. eMethods. Tissue collection, mass spectrometry, and analysis. eTable 1. Complete table of proteins identified in the human choroid‐RPE using LC‐ MS/MS. eTable 2. Top 50 signaling pathways in the human choroid‐RPE using MetaCore. eTable 3. Top 50 differentially expressed signaling pathways in the human choroid‐RPE using MetaCore. eTable 4. Differentially expressed proteins in the fovea, macula, and periphery of the human choroid‐RPE. eTable 5. Differentially expressed transcription proteins were identified in foveal, macular, and peripheral choroid‐RPE (p<0.05). eTable 6. Complement proteins identified in the human choroid‐RPE. eTable 7. Proteins associated with age related macular degeneration (AMD). This supplementary material has been provided by the authors to give readers additional information about their work. © 2014 American Medical Association. All rights reserved. 1 Downloaded From: https://jamanetwork.com/ on 09/25/2021 eFigure 1. Choroid–retinal pigment epithelial (RPE) proteomic analysis pipeline. A. The human choroid‐RPE was dissected into fovea, macula, and periphery samples. B. Fractions of proteins were isolated and digested. C. The peptide fragments were analyzed using multi‐dimensional LC‐MS/MS. D. X!Hunter, X!!Tandem, and OMSSA were used for peptide fragment identification. E. Proteins were further analyzed using bioinformatics.