An Exploration of Basic Processes for Aqueous Electrochemical Production of Hydrogen from Biomass Derived Molecules
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pulse Voltammetry Software Brochure
Data Analysis density. This feature is particularly useful for comparing data from electrodes of different areas. The analysis of the software data is performed in the Echem Analyst. Specific analysis routines have been created to Baseline Add: Baselines can be added to the data graph by either drawing a Freehand Line or by extrapolating a handle this software data files. The general features of the Echem Analyst are described in a separate brochure entitled part of the baseline with the Linear Fit feature. Redefining Electrochemical Measurement “Overview of Gamry Software.” Integrate: Integration of the current in Differential Pulse These specific routines include: Voltammetry and Square Wave Voltammetry is possible by defining a baseline and then selecting the portion of the Pulse Voltammetry Software Peak Find: Use the Region Selector button to select a curve you want to integrate. Then select Integrate from the portion of the curve that includes the region where the drop-down menu and the result is reported on the curve The Pulse Voltammetry Software adds Differential Pulse peak is located. Click on the Peak Find button to find the and also on a new tab. This software incorporates the following pulse techniques: peak position and the peak height. A perpendicular line is Voltammetry, Square Wave Voltammetry, and other drawn on the chart from the peak to the baseline. Background Subtract: A background file can be recognized pulse voltammetry techniques to the Gamry ● Square Wave subtracted from the current active data file by selecting software product family. For qualitative and mechanistic ● Square Wave Stripping Subtract from the menu and choosing the file. -

Supporting Information Redox-Active and Semi-Conducting Donor
Electronic Supplementary Material (ESI) for Journal of Materials Chemistry A. This journal is © The Royal Society of Chemistry 2018 Supporting Information Redox-active and Semi-conducting Donor-Acceptor Conjugated Microporous Polymers as Metal-free ORR Catalysts Syamantak Roy,a Arkamita Bandyopadhyay,b Mrinmay Das,c Partha Pratim Ray,c Swapan K. Patib and Tapas Kumar Maji*a a Molecular Materials Laboratory, Chemistry and Physics of Materials Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore-560064, India b New Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore- 560064 c Department of Physics, Jadavpur University, Kolkata -700032 Table of Contents Physical Measurements ..................................................................................................................2 Computational Details ....................................................................................................................3 Electrochemical Measurements ......................................................................................................3 Experimental Section......................................................................................................................5 IR Spectra .......................................................................................................................................7 PXRD .............................................................................................................................................7 -

Walljet Electrochemistry: Quantifying Molecular Transport Through Metallopolymeric and Zirconium Phosphonate Assembled Porphyrin Square Thin Films
4422 Langmuir 2004, 20, 4422-4429 Walljet Electrochemistry: Quantifying Molecular Transport through Metallopolymeric and Zirconium Phosphonate Assembled Porphyrin Square Thin Films Aaron M. Massari, Richard W. Gurney, Craig P. Schwartz, SonBinh T. Nguyen, and Joseph T. Hupp* Department of Chemistry and the Institute for Nanotechnology, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208-3113 Received January 12, 2004. In Final Form: March 7, 2004 By employing redox-active probes, condensed-phase molecular transport through nanoporous thin films can often be measured electrochemically. Certain kinds of electrode materials (e.g. conductive glass) are difficult to fabricate as rotatable disks or as ultramicroelectrodessthe configurations most often used for electrochemical permeation measurements. These limitations point to the need for a more materials- general measurement method. Herein, we report the application of walljet electrochemistry to the study of molecular transport through model metallopolymeric films on indium tin oxide electrodes. A quantitative expression is presented that describes the transport-limited current at the walljet electrode in terms of mass transport through solution and permeation through the film phase. A comparison of the film permeabilities for a series of redox probes measured using the walljet electrode and a rotating disk electrode establishes the accuracy of the walljet method, while also demonstrating similar precision for the two methods. We apply this technique to a system consisting of zirconium phosphonate assembled films of a porphyrinic molecular square. Transport through films comprising three or more layers is free from significant contributions from pinhole defects. Surprisingly, transport through films of this kind is 2-3 orders of magnitude slower than through films constructed via interfacial polymerization of nearly identical supramolecular square building blocks (Keefe; et al. -

CFD Approach to Investigating Electrochemical Hydrodynamics and Mass Transport in a Copper Electrodeposition Process Using a Rotating Disk Electrode
Int. J. Electrochem. Sci., 16 (2021) Article ID: 210239, doi: 10.20964/2021.02.14 International Journal of ELECTROCHEMICAL SCIENCE www.electrochemsci.org CFD Approach to Investigating Electrochemical Hydrodynamics and Mass Transport in a Copper Electrodeposition Process Using a Rotating Disk Electrode K.R. Kim*, H.S. Im, H.O. Nam, and S.K. Kim Korea Atomic Energy Research Institute, 1045 Daedeok-daero, Yuseong-gu, Daejeon, 305-353 Korea *E-mail: [email protected] Received: 2 September 2020 / Accepted: 10 November 2020 / Published: 31 December 2020 Three-dimensional electrochemical mass transport and concentration gradient distributions on a rotating disk electrode (RDE) were simulated in a copper electrodeposition system. This approach is based on solving the electrochemical reaction kinetics under diffusion-controlled mass transport using the Computational Fluid Dynamics (CFD) coupling technique. Diffusive boundary layer thicknesses were locally derived in association with the concentration gradient under a galvanostatic electrolysis. The simulated average diffusion boundary layer thicknesses over the electrode surface were well matched with those from the Levich correlation. Keywords: Rotating Disk Electrode, Copper Electrodeposition, Electrochemical Hydrodynamics, Mass Transport, CFD 1. INTRODUCTION An electrochemical hydrodynamic technique using a rotating disk electrode (RDE), as an electroanalytical tool, has been widely used in experimental electrochemistry for the investigation of electrode reaction kinetics under controlled mass transport conditions [1-3]. The RDE in laboratory- scale reactors provides well-defined electrolyte hydrodynamics and supports various fundamental research on material electrodeposition characteristics. The RDE system has found a wide variety of applications in investigating the designs of commercial-scaled systems. Mass transport at a rotating disk provides an experimental reproducibility to easily control the rate at which an electroactive species arrives at the electrode surface. -

EIS at a Rotating Disc Electrode
Electrochemical Impedance at a rotating disk electrode Perspective and goal • Perspective: – Researcher interested in using EIS at RDEs • Basic echem understanding • Some EIS exposure • Goal: – Provide an introduction and practical framework to run EIS at RDEs. 2 Topics • Steady state mass transfer • RDE basics and the Levich equation • Nernst diffusion layer (NDL) and the porous bounded Warburg Application • Electroactive species in biofilms • Impedance behavior of electrochemically active biofilms (EAB) at an RDE 3 Why use an RDE? • Achieve the steady state mass transfer condition: • i = nFA(D/δ)(Cbulk – Csurface) • iL = nFA(D/δ)(Cbulk) where Csurface is zero – Current is limited by the mass transfer rate – Electrode potential is predicted by the Nernst equation – “steady state” = does not change with time • Difficult to achieve in a stationary system: • δ(t) ~ (Dt)1/2 – t is time elapsed after a large potential step – Derived from the Cottrell equation 4 Source: Bard and Faulkner, "Electrochemical Methods", Chapter 1, Wiley, 2001. Difficulty with stationary systems • Background convection impacts the current measurement – After t > 20s, becomes more pronounced – Thermal gradients, vibration, other external factors • The transient current behavior and background convection influence conflicts with the EIS stability requirement, especially when wanting to run EIS at a non-zero DC current • EIS generally limited to running at the open circuit potential for diffusion-limited stationary systems 5 Source: Bard and Faulkner, "Electrochemical -

Introduction to Pulsed Voltammetric Techniques: DPV, NPV and SWV I
EC-Lab – Application Note #67 04/2019 Introduction to pulsed voltammetric techniques: DPV, NPV and SWV I – INTRODUCTION (NPV, DPV, SWV) voltammetric techniques are The pulse voltammetric techniques are compared. electroanalytical techniques mainly used to detect species of very small concentrations. II – THEORETICAL DESCRIPTION (10-6 to 10-9 mol L-1). They were developed to At low concentration, the measured current is improve voltammetric polarography expe- mainly constituted by capacitive current. The riments, in particular by minimizing the intrinsic characteristics of the pulsed capacitive (charging) current and maximizing techniques allow the user to improve the the faradaic current. detection process, for example the detection The polarography was invented by Prof. limit (DL) can reach 10 nmol L-1. Indeed, the Heyrovský (for which he won a Nobel prize) faradaic current IF Eq. (1) decreases more and consists in using a droplet of mercury as slowly than the capacitive current IC Eq. (2), an electrode, that grows, falls and is renewed. the subtraction (Fig. 1) of the current just The main advantages of using a mercury drop before and after the potential pulse (some mV electrode are that i) its surface and the during some ms) gives mainly the faradaic diffusion layer are constantly renewed, and current. not modified by deposited material during The faradaic current is given by the following electrochemical processes and ii) the proton equation [2]: reduction on mercury occurs at very high = F (1) cathodic overpotentials. Thus, it is possible to observe reactions occurring at large potential With n the Fnumber of� electrons involved in values. -

Needs Origalys Electrochem
2018 Product Catalog The OrigaLys’ solutions fit your needs OrigaLys ElectroChem SAS More than 30 years’ know-How Instruments for Electrochemical Analytical Systems ANS YEARS5 AÑOS The Company The OrigaLys’ team OrigaLys was founded in 2010, by R&D Engineers coming from Tacussel and Radiometer Analytical based in Lyon, France. Our goal is to propose to the customer "design" products with an affordable price and a high level of quality. With more than 30 years’ background and knowledge in electrochemistry and a worldwide distributor network, the company is able to provide a complete range of Instruments: Potentiostat, Galvanostat, Impedancemeter, Multi-Channels, Rotating Disk Electrode, Software, Electrodes and other Accessories (tips, pellets, corrosion cells and so on). All our products are MADE IN FRANCE. As former designers of VoltaLab devices, such as PGZ301, PGP201 or EDI101, we are able to give you a free repair diagnostic of all your instruments from Radiometer Analytical and Tacussel. 2 Worldwide Network Contact Information Distributors Headquarters Europe (13): Czech Republic, Denmark, Finland, Germany, Italy, Lithuania, Les Verchères 2 Norway, Poland, Portugal, Romania, 1er étage Spain, Sweden and The United Kingdom. 62A, avenue de l’Europe Africa (5): Algeria, Cameroon, Egypt, 69140 Rillieux-la-Pape Morocco and Tunisia. FRANCE Asia (9): Bangladesh, China, India, Iran, Malaysia, Pakistan, the Kingdom of Saudi Phone: +33 9 54 17 56 03 Arabia, Turkey and the UAE. Fax: +33 9 59 17 56 03 America (8): Argentina, Bolivia, Brazil, Email: [email protected] Canada, Colombia, Chile, Mexico and the Web site: www.origalys.com USA. If your country is not listed here. -

Part MF-9006 INSTRUCTION MANUAL Rotating Disk Electrode
RDE-2 June 2000 Part MF-9006 INSTRUCTION MANUAL Rotating Disk Electrode Bioanalytical Systems, Inc. 2701 Kent Avenue West Lafayette Indiana 47906 Copyright 2000 Bioanalytical Systems, Inc. This instrument, either wholly or in part, is manufactured for research purposes only. Use for medical diagnosis is not intended, implied, or recommended by the manufacturer. Use for this purpose and accountability for same rest entirely with the user. Table of Contents Table of Contents Section 1. Introduction . ............................................ 1 Features . ..................................................... 1 Specifications and Technical Data ..................................... 2 Accessories ..................................................... 2 Section 2. General Information ......................................... 3 User Updates ................................................... 3 Technical Changes ............................................... 3 Shipping Damage . ............................................... 3 Limited Warranty . .............................................. 3 Service....................................................... 4 Section 3. Installation . ............................................. 5 Inspection of Shipment . ......................................... 5 Parts of RDE-2. ................................................. 5 Location of RDE-2 ............................................... 5 Power Requirements ............................................. 6 Back Panel Controls and Connections ................................ -

Investigating the Accuracy of the Diffusion Coefficient Estimation
Investigating the accuracy of the diffusion coefficient estimation based on the rotating ring-disk electrode transient measurements in the cases of kinetic- limited regime and disk surface passivation. Artem V. Sergeev1*, Alexander V. Chertovich2,1 1 Physics Department, Lomonosov Moscow State University, Moscow, 119991 Russia 2 Semenov Institute of Chemical Physics, Moscow, 119991 Russia * Corresponding author. Physics Department, Lomonosov Moscow State University, Leninskie Gory, Moscow, 119991 Russia. Tel.: +7 495 939 4013. E-mail address: [email protected] Abstract The chronoamperometric measurements performed with rotating ring-disc electrode become a one of the standard techniques for the diffusion coefficient determination. However, the theoretical estimation of the transit time on which the technique is based is not strictly connected to the apparent time interval measured in the experiments. Therefore, we employed numerical simulation of RRDE in order to predict possible error of the diffusion coefficient estimation in respect to the disc electrode conditions. According to the results the obtained estimation can vary up to two times depending on the disk electrode potential. On the other hand, even an intense disk passivation process leading to the peak-like shape of the ring current transient does not affect significantly the apparent transit time and the diffusion coefficient estimation. Additionally, it was showed, that the presence of a linear segment in the disk current transient indicates that the time it took to cover most of the disk electrode with the passivation film is close to the RRDE characteristic time. Keywords RRDE, diffusion coefficient, passivation, numerical simulation Introduction A rotating ring-disk electrode (RRDE) originally was invented by Frumkin and Nekrasov1 almost half a century ago. -

Probing Fuel Cell Electrocatalyst Properties with Rotating Disk And
Document #: DRA10052 (REV003 | MAY 2016) Probing Fuel Cell Electrocatalyst Properties with Rotating Disk and Rotating Ring-Disk Electrodes Overview of the Nafion® Thin Film RRDE Technique for Oxygen Reduction Reaction This technical note discusses some practical aspects of using rotating electrodes to study fuel cell electrocatalyst materials. Specifically, a method utilizing Nafion® to immobilize platinum-bearing catalysts on electrode surfaces is described. This approach has been successfully employed by many researchers to probe both the (desired) kinetic and (undesired) peroxide generating properties of electrocatalysts used for the reduction of dioxygen. 1. Introduction Recent trends in fuel cell research and production have increased the demand for fundamental information about the behavior of fuel cell electrocatalysts. Particular attention to those catalysts involved in the oxygen reduction reaction (ORR) has been driven by interest in / fuel cells based on polymer electrolyte membranes. [1] Because ORR 2 2 electrocatalysts typically make use of very expensive metal particles (most notably, nanoscale structures involving platinum), a key issue in fuel cell research is the search for lower cost catalyst materials. Figure 1. ChangeDisk Rotating Ring-Disk Electrode. Investigators from a wide variety of backgrounds are bringing their efforts to bear on this economic issue, developing catalytic materials with less noble metal content (i.e., a dilution approach) and/or a more efficient use of the existing noble metal content (i.e., engineering particle morphology). Regardless of the approach taken, these researchers generally end up producing a large number of candidate ORR catalysts which must be screened. The two screening criteria of most interest are the ORR kinetics (which should be as fast as possible) and the amount of peroxide formed, which should be as low as possible. -

Article1379421186 Mariame Et Al.Pdf
Vol. 7(9), pp. 318-324, Sept, 2013 DOI :10.5897/AJPAC2013. 0516 African Journal of Pure and Applied ISSN 1996 - 0840 © 2013 Academic Journals Chemistry http://www.academicjournals.org/AJPAC Full Length Research Paper Assessment of dissolved manganese (II) pollution in river water by differential pulse cathodic stripping voltammetry: A case study of River Boubo, Côte d'Ivoire Coulibaly Mariame1, 2*, Yao N’Guessan Alfred2, Bamba Drissa3, Ghanjaoui Mohammed El Amine1, Yousry M. Issa4 and El Rhazi Mamia1 1Laboratoire d’Electrochimie et de Chimie Physique Faculté des Sciences et Techniques BP146, Mohammedia, Maroc. 2Laboratoire des Sciences Physiques Fondamentales et Appliquées, Ecole Normale Supérieure d’Abidjan 08 BP 10 Abidjan 08 Côte d’Ivoire. 3Laboratoire de Chimie des Eaux, Ecole Normale Supérieure d’Abidjan, 08 BP 10 Abidjan 08 Côte d’Ivoire. 4Department of Chemistry, Faculty of Science, Cairo University, Giza, Egypt. Accepted 21 August 2013 Boubo river and lagoon, south Côte d’Ivoire, are contaminated by manganese released in wastewater from manganese exploitation and from the soil of Grand-Lahou region in Côte d’Ivoire. So, there is a need to develop a simple method for the evaluation of manganese present in natural water. This work reports the utility of rapid and inexpensive method for determination of dissolved manganese (II) in natural water by cathodic stripping voltammetry using rotating carbon paste disk electrode. The experimental parameters for the determination of manganese (II) were optimized. Under the optimum conditions, the method was successfully applied to determination of free manganese in river and lagoon samples taken near the manganese exploitation in the area of Grand-Lahou, Côte d’Ivoire. -
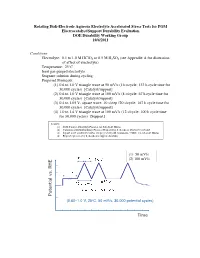
Rotating Disk-Electrode Aqueous Electrolyte Accelerated Stress Tests for PGM Electrocatalyst/Support Durability Evaluation DOE Durability Working Group 10/4/2011
Rotating Disk-Electrode Aqueous Electrolyte Accelerated Stress Tests for PGM Electrocatalyst/Support Durability Evaluation DOE Durability Working Group 10/4/2011 Conditions Electrolyte: 0.1 to 1.0 M HClO4 or 0.5 M H2SO4 (see Appendix A for discussion of effect of electrolyte) Temperature: 25°C Inert gas-purged electrolyte Stagnant solution during cycling Proposed Protocols: (1) 0.6 to 1.0 V triangle wave at 50 mV/s (16 s/cycle; 133 h cycle time for 30,000 cycles) [Catalyst/support] (2) 0.6 to 1.0 V triangle wave at 100 mV/s (8 s/cycle; 67 h cycle time for 30,000 cycles) [Catalyst/support] (3) 0.4 to 1.05 V, square wave, 10 s/step (20 s/cycle; 167 h cycle time for 30,000 cycles) [Catalyst/support] (4) 1.0 to 1.6 V triangle wave at 100 mV/s (12 s/cycle; 100 h cycle time for 30,000 cycles) [Support] Sources: (1) DOE Catalyst Durability Protocol for Sub-Scale MEAs (2) Variation of DOE Durability Protocol Proposed by S. Kocha to decrease cycle time (3) Square wave accelerated stress test proposed by M. Gummalla, UTRC, for sub-scale MEAs (4) Proposed protocol by S. Kocha for support durability (1) 50 mV/s 8 s 8 s 1.0 V (2) 100 mV/s 30 s 16 s/cycle 0.60 V Scan Rate : 50 mV/s Potential Potential RHE vs. (0.60–1.0 V, 25oC, 50 mV/s, 30,000 potential cycles) Time (3) 6 s 6 s 1.6 V (4) 30 s 12 s/cycle 1.0 V Scan Rate : 100 mV/s Potential Potential RHEvs.