Freestyle 1.4 – User Guide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ACE-2019-Query-Builder-And-Tree
Copyright © 2019 by Aras Corporation. This material may be distributed only subject to the terms and conditions set forth in the Open Publication License, V1.0 or later (the latest version is presently available at http://www.opencontent.org/openpub/). Distribution of substantively modified versions of this document is prohibited without the explicit permission of the copyright holder. Distribution of the work or derivative of the work in any standard (paper) book form for a commercial purpose is prohibited unless prior permission is obtained from the copyright holder. Aras Innovator, Aras, and the Aras Corp "A" logo are registered trademarks of Aras Corporation in the United States and other countries. All other trademarks referenced herein are the property of their respective owners. Microsoft, Office, SQL Server, IIS and Windows are either registered trademarks or trademarks of Microsoft Corporation in the United States and/or other countries. Notice of Liability The information contained in this document is distributed on an "As Is" basis, without warranty of any kind, express or implied, including, but not limited to, the implied warranties of merchantability and fitness for a particular purpose or a warranty of non-infringement. Aras shall have no liability to any person or entity with respect to any loss or damage caused or alleged to be caused directly or indirectly by the information contained in this document or by the software or hardware products described herein. Copyright © 2019 by Aras Corporation. This material may be distributed only subject to the terms and conditions set forth in the Open Publication License, V1.0 or later (the latest version is presently available at http://www.opencontent.org/openpub/). -

Kurzweil 1000 Version 12 New Features
Kurzweil 1000 Version 12 New Features For the most up-to-date feature information, refer to the Readme file on the product CD. The following is a summary of what’s new in Version 12. For complete details, go to the online Manual by pressing Alt+H+O. Where applicable, Search key words are provided for you to use in the online Manual. • While you may not notice any difference, the internal structure of Kurzweil 1000 Version 12 has been overhauled and now uses Microsoft .NET Framework. The intent is to make it easier for Cambium Learning Technologies to develop features for the product going forward. • Note that Kurzweil 1000 Version 12 now supports 64-bit operating systems and Microsoft Windows 7 operating system. • As always, Kurzweil 1000 has the latest OCR engines, FineReader 9.0.1 and ScanSoft 16.2. The new ScanSoft version includes recognition languages from the Sami family. • An especially exciting new feature is the New User Wizard, a set of topics that introduces and walks new users through a number of Kurzweil 1000 features and preference setups. It appears when you start up Kurzweil 1000, but can be disabled and accessed from the Help menu by pressing Alt+H+W. (Search: New User Wizard.) • Currency Recognition has been updated to support new bills. Note that Currency Recognition now requires a color scanner. • New features and enhancements in reference tools include: 1. updates of the American Heritage Dictionary and Roget’s Thesaurus. 2. the ability to find up to 114 of your previously looked up entries; and last but not least the addition to dictionary and thesaurus lookup of human pronunciations and Anagrams. -

A Mobile Interface for Navigating Hierarchical Information Space$
Journal of Visual Languages and Computing 31 (2015) 48–69 Contents lists available at ScienceDirect Journal of Visual Languages and Computing journal homepage: www.elsevier.com/locate/jvlc A mobile interface for navigating hierarchical information space$ Abhishek P. Chhetri a,n, Kang Zhang b,c, Eakta Jain c,d a Computer Engineering Program, Erik Jonsson School of Engineering and Computer Science, University of Texas at Dallas, Richardson, TX 65080-3021, USA b School of Software Engineering, Tianjin University, Tianjin, China c Department of Computer Science, University of Texas at Dallas, Richardson, TX 65080-3021, USA d Texas Instruments, Dallas, TX, USA article info abstract Article history: This paper presents ERELT (Enhanced Radial Edgeless Tree), a tree visualization approach Received 2 June 2015 on modern mobile devices. ERELT is designed to offer a clear visualization of any tree Accepted 5 October 2015 structure with intuitive interaction. Such visualization can assist users in interacting with Available online 22 October 2015 a hierarchical structure such as a media collection, file system, etc. General terms: In the ERELT visualization, a subset of the tree is displayed at a time. The displayed tree Algorithms size depends on the maximum number of tree elements that can be put on the screen Design while maintaining clarity. Users can quickly navigate to the hidden parts of the tree Human factors through touch-based gestures. We have conducted a user study to evaluate this visuali- zation for a music collection. The study results show that this approach reduces the time Keywords: and effort in navigating tree structures for exploration and search tasks. -

Completeview™ Video Client User Manual
CompleteView™ Video Client User Manual CompleteView™ Version 4.7 Contents Introduction ................................................................................................................ 1 End User License Agreement ........................................................................................................................1 System Requirements ....................................................................................................................................3 Operation .................................................................................................................... 3 Getting Started ...............................................................................................................................................3 Starting the CompleteView Client ................................................................................................................3 Logging In to the CompleteView Client .......................................................................................................4 The Login Dialog .........................................................................................................................................4 Client Application Update ...........................................................................................................................5 Contacting Video Servers ............................................................................................................................6 Application Overview -
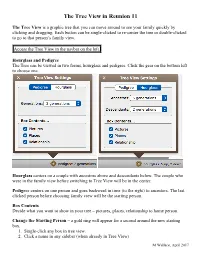
Tree View Handout
The Tree View in Reunion 11 The Tree View is a graphic tree that you can move around to see your family quickly by clicking and dragging. Each button can be single-clicked to re-center the tree or double-clicked to go to that person’s family view. Access the Tree View in the navbar on the left. Hourglass and Pedigree The Tree can be viewed in two forms, hourglass and pedigree. Click the gear on the bottom left to choose one. Hourglass centers on a couple with ancestors above and descendants below. The couple who were in the family view before switching to Tree View will be in the center. Pedigree centers on one person and goes backward in time (to the right) to ancestors. The last clicked person before choosing family view will be the starting person. Box Contents Decide what you want to show in your tree – pictures, places, relationship to home person. Change the Starting Person – a gold ring will appear for a second around the new starting box. 1. Single-click any box in tree view. 2. Click a name in any sidebar (when already in Tree View) M Wallace, April 2017 Page 2 of 2 3. Drag a person button from Family View to Tree View in navbar. 4. Drag a name from the sidebar to Tree View in navbar. 5. Control-click (right-click) any box in Tree View and choose someone from the Person menu. From this menu you can also - Mark/unmark the person - Mark everyone in this tree view - Switch back to Family View for that person - Open the Edit Person panel for that person - Show the Find Relative sidebar to re-calculate relationships for that person - Return to Starting Box – if you have moved around a lot and you want to go back to the beginning person/family, choose Edit > Locate Starting Box or Command B. -

Visualization Service Bus
Visualization Service Bus Abstract— In this research, we are applying modern Service-Oriented Architecture (SOA) technologies to make complex visualizations realizable without intensive graphics programming; in fact, without writing code at all. A Visualization Service Bus is the integration of two powerful Extensible Markup Language (XML) technologies, Extensible 3D (X3D) and an Enterprise Service Bus (ESB), which allows users to integrate data and develop user defined visualizations together. Analysts use graphical interfaces to construct visual elements, assemble a dynamic scene, connect to and transform data from a variety of sources, and map scientific data to the scene. The development and implementation of this visualization architecture allows non-programmers to develop their own visualization applications for their specific needs. Index Terms—X3D, XML, ESB, BPEL, XSLT, XSD, SOA, JBI, JMS, and XPATH. 1 INTRODUCTION In the aviation research community, scientists analyze, mine, verify, manipulate and visualize data in many ways to understand complex 1.2 Binding Components problems and to convey research results. Analysts today use a wide Binding components communicate with external services. These variety of visualization tools to demonstrate their research findings. components access services over a known protocol and data format. Typically, these tools are designed to read in a specific type of data Examples include HTTP, SOAP, JMS, TCP, FTP, and SMTP. The in a proprietary format and transport protocol. These visualizations binding component is responsible for communicating with external tend to be inherently rigid, and large software efforts are needed to services over a specified protocol and converting that data into the make changes or implement new features. -
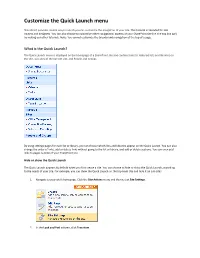
Customize the Quick Launch Menu
Customize the Quick Launch menu SharePoint provides several ways in which you can customize the navigation of your site. This tutorial is intended for site owners and designers. You can also choose to customize other navigational aspects of your SharePoint site (i.e. the top link bar) by visiting our other tutorials. Note: You cannot customize the breadcrumb navigation at the top of a page. What is the Quick Launch? The Quick Launch menu is displayed on the homepage of a SharePoint site and contains links to featured lists and libraries on the site, sub-sites of the current site, and People and Groups. By using settings pages for each list or library, you can choose which lists and libraries appear on the Quick Launch. You can also change the order of links, add or delete links without going to the list or library, and add or delete sections. You can even add links to pages outside of your SharePoint site. Hide or show the Quick Launch The Quick Launch appears by default when you first create a site. You can choose to hide or show the Quick Launch, according to the needs of your site. For example, you can show the Quick Launch on the top-level site and hide it on sub-sites. 1. Navigate to your site’s homepage. Click the Site Actions menu and then select Site Settings. 2. In the Look and Feel column, click Tree view. 3. To hide the Quick Launch, deselect the Enable Quick Launch checkbox. To show the Quick Launch, select the Enable Quick Launch checkbox. -

Branching out with the Tree View Control in SAS/AF® Software Lynn Curley, SAS Institute Inc., Cary, NC Scott Jackson, SAS Institute Inc., Cary, NC
SAS Global Forum 2008 Applications Development Paper 019-2008 Branching Out with the Tree View Control in SAS/AF® Software Lynn Curley, SAS Institute Inc., Cary, NC Scott Jackson, SAS Institute Inc., Cary, NC ABSTRACT Harness the power of the Tree View Control in SAS/AF® software to present hierarchical data like the Microsoft Windows Explorer folder list. The Tree View Control was previously experimental, but has been promoted to production status in SAS® 9.2 thanks to customer feedback. Through examples, this paper introduces you to both the Tree View and Tree Node classes and details the attributes and methods that help make the Tree View a customizable, multi-purpose object. INTRODUCTION Several years ago, SAS redirected development resources from SAS/AF software toward SAS® AppDev Studio, which is based on a Java development environment. At that time, SAS encouraged its customers to move in this direction. The migration led some SAS customers to question the future of SAS/AF software. Although SAS is still committed to developing technologies that are based on Java, SAS recognizes that many of its customers want and need the rich object-oriented programming environment that SAS/AF software offers. Therefore, in response to customer feedback, there are several enhancements for SAS/AF software in SAS 9.2, including promotion of the experimental Tree View and Tree Node Controls to production status. This paper discusses the capabilities of the Tree View Control in SAS/AF software and meets the following objectives: • details the hierarchical structure of a Tree View Control, including the node objects that populate the tree view • demonstrates two methods to search for nodes in a Tree View Control • includes a customizable example of using a Tree View Control as a menu via drag-and-drop communication. -

Xcalibur 3.1 Qual Browser User Guide Version A
Thermo Xcalibur Qual Browser User Guide Software Version 3.1 XCALI-97617 Revision A August 2014 © 2014 Thermo Fisher Scientific Inc. All rights reserved. Accela, Xcalibur, and Q Exactive are registered trademarks of Thermo Fisher Scientific Inc. in the United States. Microsoft, Windows, and Excel are registered trademarks of Microsoft Corporation in the United States and other countries. Adobe, Acrobat, and Reader are registered trademarks of Adobe Systems Incorporated in the United States and other countries. All other trademarks are the property of Thermo Fisher Scientific Inc. and its subsidiaries. Thermo Fisher Scientific Inc. provides this document to its customers with a product purchase to use in the product operation. This document is copyright protected and any reproduction of the whole or any part of this document is strictly prohibited, except with the written authorization of Thermo Fisher Scientific Inc. The contents of this document are subject to change without notice. All technical information in this document is for reference purposes only. System configurations and specifications in this document supersede all previous information received by the purchaser. This document is not part of any sales contract between Thermo Fisher Scientific Inc. and a purchaser. This document shall in no way govern or modify any Terms and Conditions of Sale, which Terms and Conditions of Sale shall govern all conflicting information between the two documents. Release history: Revision A, August 2014 Software version: Xcalibur 3.1 and later For Research Use Only. Not for use in diagnostic procedures. C Contents Preface . ix About This Guide. .ix Related Documentation . x Safety and Special Notices . -

Graph, Tree and DAG Object and Window Structures
COMP 401 Prasun Dewan Graph, Tree and DAG Object and Window Structures In the previous chapter, we learnt how to create complex logical structures included structured and composite objects and shapes. In this chapter, we will classify these structured into three important categories of increasing complexity: trees, DAGS, and graphs. In later courses, you will theoretically analyze these structured in great depth. Here, we will see some of the practical consequences, of (deliberately or accidentally) choosing among these categories. We will redo the Cartesian plane example to illustrate the structures and consequences. Cartesian Plane with Composite Line Objects Recall that we have created two different interfaces for a geometric line. One, called Line, was both an atomic shape and atomic object – all properties were primitive. public interface Line { public int getX(); public void setX(int newX); public int getY(); public void setY(int newY); … } The other, LineWithObjectProperty, was also an atomic shape but a composite object – its location was an object. public interface LineWithObjectProperty { public Point getLocation(); public void setLocation(Point newLocation); … } In our previous CartesianPlane solution, we embedded two Line objects to represent the two axes: public interface CartesianPlane { public Line getXAxis(); public Line getYAxis(); … } Let us make the problem more interesting by representing the axes as LineWithObjectProperty and also making the locations of these two axes as properties of the plane. public interface -

JAWS® for Windows® Quick Start Guide
JAWS® for Windows® Quick Start Guide Freedom Scientific, Inc. PUBLISHED BY Freedom Scientific www.FreedomScientific.com Information in this document is subject to change without notice. No part of this publication may be reproduced or transmitted in any form or any means electronic or mechanical, for any purpose, without the express written permission of Freedom Scientific. Copyright © 2018 Freedom Scientific, Inc. All Rights Reserved. JAWS is a registered trademark of Freedom Scientific, Inc. in the United States and other countries. Microsoft, Windows 10, Windows 8.1, Windows 7, and Windows Server are registered trademarks of Microsoft Corporation in the U.S. and/or other countries. Sentinel® is a registered trademark of SafeNet, Inc. ii Table of Contents Welcome to JAWS for Windows ................................................................. 1 System Requirements ............................................................................... 2 Installing JAWS ............................................................................................ 3 Activating JAWS ....................................................................................... 3 Dongle Authorization ................................................................................. 4 Network JAWS .......................................................................................... 5 Running JAWS Startup Wizard ................................................................. 5 Installing Vocalizer Expressive Voices ..................................................... -

Administration of the LOC Net Platform
17 June 2015 Page 1 of 48 8G, The Leathermarket, Weston Street, London, SE1 3ER 0203 176 5380 www.trilliumsystems.net [email protected] How to Guide Administration of the LOC Net platform Attention: Local Optical Committee website Admins Company: LOCSU Prepared By: Trillium Systems Date: 17 June 2015 Reference: LOCSU-TM-001 Version: 1.1 Please Note: All information contained in this document is copyright Trillium Systems Limited 2015. The information is intended for the recipient, Local Optical Comittee administrators, and their associated companies and agents. The intellectual property contained herein remains the property of Trillium Systems Limited and may not be used, applied, distributed, referenced, forwarded or adapted in any way without the written consent of Trillium Systems Limited. Breach of this intellectual copyright may result in legal action. All Trademarks are property of their respective owners. All rights reserved. Copyright Trillium Systems Limited 2015. Private and Confidential 17 June 2015 Page 2 of 48 Table of Contents The LOC Net platform................................................................................................................... 6 Overview ............................................................................................................................................. 6 Concepts & Terminology .............................................................................................................. 7 Planning your LOC Net Site ..........................................................................................................