Nuclear RNA Concentration Coordinates RNA Production with Cell Size in Human Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Autophagy: from Basic Science to Clinical Application
nature publishing group REVIEW See COMMENTARY page XX Autophagy: from basic science to clinical application J Va n L i m b e r g e n 1 , 2 , 3 , C S t e v e n s 4 , E R N i m m o 1 , D C W i l s o n 2 , 3 a n d J S a t s a n g i 1 Autophagy is a cellular pathway involved in protein and organelle degradation, which is likely to represent an innate adaptation to starvation. In times of nutrient deficiency, the cell can self-digest and recycle some nonessential components through nonselective autophagy, thus sustaining minimal growth requirements until a food source becomes available. Over recent years, autophagy has been implicated in an increasing number of clinical scenarios, notably infectious diseases, cancer, neurodegenerative diseases, and autoimmunity. The recent identification of the importance of autophagy genes in the genetic susceptibility to Crohn ’ s disease suggests that a selective autophagic response may play a crucial role in the pathogenesis of common complex immune-mediated diseases. In this review, we discuss the autophagic mechanisms, their molecular regulation, and summarize their clinical relevance. This progress has led to great interest in the therapeutic potential of manipulation of both selective and nonselective autophagy in established disease. INTRODUCTION The ability to adapt to environmental change is essential for sur- Autophagy encompasses several distinct processes involving vival. This is true for the organism as a whole and for individual the delivery of portions of the cytoplasm to the lysosome for cells alike. -

XPB Induces C1D Expression to Counteract UV-Induced Apoptosis
Published OnlineFirst June 8, 2010; DOI: 10.1158/1541-7786.MCR-09-0467 Molecular DNA Damage and Cellular Stress Responses Cancer Research XPB Induces C1D Expression to Counteract UV-Induced Apoptosis Guang Li1, Juhong Liu2, Mones Abu-Asab1, Shibuya Masabumi3, and Yoshiro Maru4 Abstract Although C1D has been shown to be involved in DNA double-strand break repair, how C1D expression was induced and the mechanism(s) by which C1D facilitates DNA repair in mammalian cells remain poorly understood. We and others have previously shown that expression of xeroderma pigmentosum B (XPB) pro- tein efficiently compensated the UV irradiation–sensitive phenotype of 27-1 cells, which lack functional XPB. To further explore XPB-regulated genes that could be involved in UV-induced DNA repair, differential dis- play analysis of mRNA levels from CHO-9, 27-1, and 27-1 complemented with wild-type XPB was done and C1D gene was identified as one of the major genes whose expression was significantly upregulated by restoring XPB function. We found that XPB is essential to induce C1D transcription after UV irradiation. The increase in C1D expression effectively compensates for the UV-induced proteolysis of C1D and thus maintains cellular C1D level to cope with DNA damage inflicted by UV irradiation. We further showed that although insufficient to rescue 27-1 cells from UV-induced apoptosis by itself, C1D facilitates XPB DNA repair through direct interaction with XPB. Our findings provided direct evidence that C1D is associated with DNA repair complex and may promote repair of UV-induced DNA damage. Mol Cancer Res; 8(6); 885–95. -

Oncogenic H-Ras Up-Regulates Expression of ERCC1 to Protect Cells from Platinum-Based Anticancer Agents
[CANCER RESEARCH 64, 4849–4857, July 15, 2004] Oncogenic H-Ras Up-Regulates Expression of ERCC1 to Protect Cells from Platinum-Based Anticancer Agents Cha-Kyung Youn,1 Mi-Hwa Kim,1 Hyun-Ju Cho,1 Hong-Beum Kim,1 In-Youb Chang,1 Myung-Hee Chung,3 and Ho Jin You1,2 1Research Center for Proteineous Materials and 2Department of Pharmacology, School of Medicine, Chosun University, Gwangju, and 3Department of Pharmacology, School of Medicine, Seoul National University, Seoul, Korea ABSTRACT elucidated, evidence suggests that the activated Ras may contribute to cisplatin resistance by stimulating the DNA repair activity (9, 12, 13). Tumors frequently contain mutations in the ras genes, resulting in the Hence, there has been considerable interest in determining which constitutive activation of the Ras-activated signaling pathway. The acti- vation of Ras is involved not only in tumor progression but also in the proteins mediate the altered DNA repair capacity in activated Ras- development of resistance of the tumor cells to platinum-based chemo- containing cells. However, the downstream target genes of the onco- therapeutic agents. To investigate the potential mechanisms underlying genic Ras, which are involved in the enhancement of the DNA repair this resistance, we analyzed the effect of activated H-Ras on the expression activity, are unclear. of the nucleotide excision repair genes. Here we identified ERCC1, which Cisplatin is one of the most effective and widely used anticancer is one of the key enzymes involved in nucleotide excision repair, as being drugs for treating human solid tumors (14). However, its therapeutic markedly up-regulated by the activated H-Ras. -

Characterization of Gf a Drosophila Trimeric G Protein Alpha Subunit
Characterization of Gf a Drosophila trimeric G protein alpha subunit Naureen Quibria Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2012 © 2012 Naureen Quibria All rights reserved Abstract Characterization of Gf a Drosophila trimeric G-protein alpha subunit Naureen Quibria In the morphogenesis of tissue development, how coordination of patterning and growth achieve the correct organ size and shape is a principal question in biology. Efficient orchestrating mechanisms are required to achieve this and cells have developed sophisticated systems for reception and interpretation of the multitude of extracellular stimuli to which they are exposed. Plasma membrane receptors play a key role in the transmission of such signals. G-protein coupled receptors (GPCRs) are the largest class of cell surface receptors that respond to an enormous diversity of extracellular stimuli, and are critical mediators of cellular signal transduction in eukaryotic organisms. Signaling through GPCRs has been well characterized in many biological contexts. While they are a major class of signal transducers, there are not many defined instances where GPCRs have been implicated in the process of development to date. The Drosophila wing provides an ideal model system to elucidate and address the role of GPCRs in development, as its growth is regulated by a small number of conserved signaling pathways. In my thesis work, I address the role of a trimeric G alpha protein in Drosophila, Gαf, and what part it may play in development. In particular, I explore the role of Gαf as an alpha subunit of a trimeric complex, to determine what heptahelical receptors might act as its cognate receptor. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

P38 MAPK-Mediated Activation of NF-Kb by the Rhogef Domain of Bcr
Oncogene (2002) 21, 4601 – 4612 ª 2002 Nature Publishing Group All rights reserved 0950 – 9232/02 $25.00 www.nature.com/onc p38 MAPK-mediated activation of NF-kB by the RhoGEF domain of Bcr Malgorzata Korus1,2, Gwendolyn M Mahon1,2, Li Cheng1 and Ian P Whitehead*,1 1Department of Microbiology and Molecular Genetics, UMDNJ – New Jersey Medical School, Newark, New Jersey, NJ 07103, USA The oncogenic fusion protein p210 Bcr-Abl is causally et al., 1988; Fainstein et al., 1987; Konopka et al., associated with virtually all cases of chronic myelogenous 1984; Kurzrock et al., 1987; Walker et al., 1987). leukemia. The wild-type Bcr product has several Although animal models suggest that elevated levels of recognizable structural and functional motifs including Abl tyrosine kinase activity contribute to Bcr-Abl a domain that contains guanine nucleotide exchange mediated leukemogenesis (Lugo et al., 1990), the activity for Rho family GTPases (DH/PH domain). residues that distinguish the rearrangements associated Although this domain is retained within p210 Bcr-Abl, it with ALL (p185 Bcr-Abl) from the rearrangements has no known signaling activities in vivo. Here we report associated with CML (p210 Bcr-Abl) reside within the that a fragment of Bcr that encodes the isolated DH/PH Bcr protein. Thus, a complete understanding of the domain is a potent activator of the NF-kB transcription biological mechanisms that distinguish ALL from factor. Within the context of full length Bcr, this activity CML may depend upon the characterization of the is regulated by proximal flanking sequences that suppress signaling activities that reside within Bcr. -
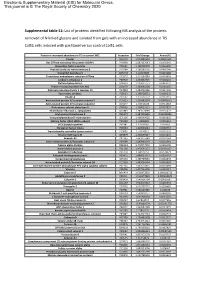
Supplemental Table S1
Electronic Supplementary Material (ESI) for Molecular Omics. This journal is © The Royal Society of Chemistry 2020 Supplemental table S1: List of proteins identified following MS analysis of the proteins removed of N-linked glycans and isolated from gels with an increased abundance in TIS Cal51 cells induced with paclitaxel versus control Cal51 cells. Protein in increased abundance in TIS vs control WCL Accession Fold Change Anova (P) Plectin Q15149 1.073855593 0.00691631 Ras GTPase-activating-like protein IQGAP1 P46940 1.087337643 0.0176342 Elongation factor1-gamma P26641 1.138709703 0.0116496 Peptidyl-prolyl cis-transisomerase B P23284 1.188383105 0.0436246 Dipeptidyl peptidase 3 Q9NY33 1.20163605 0.0215448 Transitional endoplasmic reticulum ATPase P55072 1.214194884 0.0449691 Carbonic anhydrase 2 P00918 1.232852325 0.0158141 Clathrin heavy chain 1 Q00610 1.239621773 0.0463237 Protein transport protein Sec 31A O94979 1.263565104 0.0284155 Aldo-ketoreductase family 1 member C1 Q04828 1.282092186 0.0324406 Spermidine synthase P19623 1.298728621 0.0196232 Plastin-3 P13797 1.310756772 0.0161319 Actin-related protein 2/3 complex subunit 5 O15511 1.333483524 0.00476923 Actin-related protein 2/3 complex subunit 2 O15144 1.35416168 0.0411018 Proteasome subunit alpha type-5 P28066 1.358015551 0.0337657 Thioredoxin reductase 1, cytoplasmic Q16881 1.383670089 0.0235472 Acyl-protein thioesterase 2 O95372 1.387415589 0.00233899 Isoaspartylpeptidase/L-asparaginase Q7L266 1.408149002 0.0319602 Splicing factor U2AF 65kDa subunit P26368 1.41489991 0.0256619 -

Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/Mtor Cascade Inhibitors: How Mutations Can Result in Therapy Resistance and How to Overcome Resistance
www.impactjournals.com/oncotarget/ Oncotarget, October, Vol.3, No 10 Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascade Inhibitors: How Mutations Can Result in Therapy Resistance and How to Overcome Resistance James A. McCubrey1, Linda S. Steelman1, William H. Chappell1, Stephen L. Abrams1, Richard A. Franklin1, Giuseppe Montalto2, Melchiorre Cervello3, Massimo Libra4, Saverio Candido4, Grazia Malaponte4, Maria C. Mazzarino4, Paolo Fagone4, Ferdinando Nicoletti4, Jörg Bäsecke5, Sanja Mijatovic6, Danijela Maksimovic- Ivanic6, Michele Milella7, Agostino Tafuri8, Francesca Chiarini9, Camilla Evangelisti9, Lucio Cocco10, Alberto M. Martelli9,10 1 Department of Microbiology and Immunology, Brody School of Medicine at East Carolina University, Greenville, NC, USA 2 Department of Internal Medicine and Specialties, University of Palermo, Palermo, Italy 3 Consiglio Nazionale delle Ricerche, Istituto di Biomedicina e Immunologia Molecolare “Alberto Monroy”, Palermo, Italy 4 Department of Bio-Medical Sciences, University of Catania, Catania, Italy 5 Department of Medicine, University of Göttingen, Göttingen, Germany 6 Department of Immunology, Instititue for Biological Research “Sinisa Stankovic”, University of Belgrade, Belgrade, Serbia 7 Regina Elena National Cancer Institute, Rome, Italy 8 Sapienza, University of Rome, Department of Cellular Biotechnology and Hematology, Rome, Italy 9 Institute of Molecular Genetics, National Research Council-Rizzoli Orthopedic Institute, Bologna, Italy. 10 Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy Correspondence to: James A. McCubrey, email: [email protected] Keywords: Targeted Therapy, Therapy Resistance, Cancer Stem Cells, Raf, Akt, PI3K, mTOR Received: September 12, 2012, Accepted: October 18, 2012, Published: October 20, 2012 Copyright: © McCubrey et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. -

Discovery of the Essential Roles of CRK9 in Gene Expression and Of
University of Connecticut OpenCommons@UConn Doctoral Dissertations University of Connecticut Graduate School 5-27-2015 Lessons from Trypanosome TFIIH: Discovery of the Essential Roles of CRK9 in Gene Expression and of the Divergent Helicase Paralog XPB-R in Nucleotide Excision Repair Nitika Badjatia University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/dissertations Recommended Citation Badjatia, Nitika, "Lessons from Trypanosome TFIIH: Discovery of the Essential Roles of CRK9 in Gene Expression and of the Divergent Helicase Paralog XPB-R in Nucleotide Excision Repair" (2015). Doctoral Dissertations. 866. https://opencommons.uconn.edu/dissertations/866 Lessons from Trypanosome TFIIH: Discovery of the Essential Roles of CRK9 in Gene Expression and of the Divergent Helicase Paralog XPB-R in Nucleotide Excision Repair Nitika Badjatia University of Connecticut, [2015] Eukaryotic TFIIH consists of a core of seven subunits, including the DNA helicase Xeroderma Pigmentosum B (XPB), and a cyclin-dependent kinase (CDK)-activating complex (CAK) that contains CDK7. XPB is crucial for DNA unwinding during transcription and nucleotide excision repair (NER) while the CAK complex phosphorylates RNA polymerase II (RNAPII) carboxy terminal domain (CTD), enabling the enzyme’s promoter clearance. Trypanosoma brucei, Trypanosoma cruzi and Leishmania spp. are lethal human parasites, belonging to the early-diverged order Kinetoplastida. They transcribe genes polycistronically and require spliced leader (SL) trans splicing for the maturation of mRNAs from polycistronic precursors. SL RNA gene (SLRNA) transcription by RNAPII depends on trypanosome TFIIH complex which contains orthologs of all core subunits but lacks the CAK complex. Despite this, the trypanosome CTD is phosphorylated and essential for SLRNA transcription. -

Melanomas Are Comprised of Multiple Biologically Distinct Categories
Melanomas are comprised of multiple biologically distinct categories, which differ in cell of origin, age of onset, clinical and histologic presentation, pattern of metastasis, ethnic distribution, causative role of UV radiation, predisposing germ line alterations, mutational processes, and patterns of somatic mutations. Neoplasms are initiated by gain of function mutations in one of several primary oncogenes, typically leading to benign melanocytic nevi with characteristic histologic features. The progression of nevi is restrained by multiple tumor suppressive mechanisms. Secondary genetic alterations override these barriers and promote intermediate or overtly malignant tumors along distinct progression trajectories. The current knowledge about pathogenesis, clinical, histological and genetic features of primary melanocytic neoplasms is reviewed and integrated into a taxonomic framework. THE MOLECULAR PATHOLOGY OF MELANOMA: AN INTEGRATED TAXONOMY OF MELANOCYTIC NEOPLASIA Boris C. Bastian Corresponding Author: Boris C. Bastian, M.D. Ph.D. Gerson & Barbara Bass Bakar Distinguished Professor of Cancer Biology Departments of Dermatology and Pathology University of California, San Francisco UCSF Cardiovascular Research Institute 555 Mission Bay Blvd South Box 3118, Room 252K San Francisco, CA 94158-9001 [email protected] Key words: Genetics Pathogenesis Classification Mutation Nevi Table of Contents Molecular pathogenesis of melanocytic neoplasia .................................................... 1 Classification of melanocytic neoplasms -

University of Florida Thesis Or Dissertation Formatting
AAA ATPASE CDC48A AND ITS ASSOCIATION WITH UBIQUITIN-LIKE SAMP1 AND DNA REPAIR IN ARCHAEA By SWATHI DANTULURI A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF FLORIDA 2018 © 2018 Swathi Dantuluri I dedicate this to my family and friends for their endless love, support and company.Everybody who has taken the time and energy to teach me everything small or big. ACKNOWLEDGMENTS During my graduate career at the University of Florida, there are many people to whom I will be eternally indebted to for supporting and advising me along the way. Firstly, I would like to thank my advisor, Dr. Julie A. Maupin-Furlow for providing me the opportunity to work under her guidance as well as for all her help, advice, support, and encouragement throughout my time in her lab. I would also like to thank my committee members Dr. Sixue Chen, Dr.Tony Romeo and Dr. Nemat.O.Keyhani for their constructive advice and guidance. I must also express my gratitude to all the past and current Maupin lab members with whom I developed a warm and friendly working relationship over time, especially Dr. Nathaniel Hepowit who patiently taught me all the lab techniques, Dr. Xian Fu, Dr. Lana J. McMillan, Dr. Shiyun Cao, Dr. Sungmin Hwang, Zachary Adams, Cuper Ramirez, Paula Mondragon, and Jou Chin Chan. I also would like to thank all the undergraduate researchers in our lab for their dedication: Shae Marguiles, Sonam Parag, Gayathri Srinivasan, and Whinkie Leung. -

The Small Gtpase Rheb Affects Central Brain Neuronal Morphology and Memory Formation in Drosophila
The Small GTPase Rheb Affects Central Brain Neuronal Morphology and Memory Formation in Drosophila Heather L. D. Brown1¤a, Karla R. Kaun2¤b, Bruce A. Edgar1*¤c 1 Basic Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, Washington, United States of America, 2 Department of Anatomy, University of California San Francisco, San Francisco, California, United States of America Abstract Mutations in either of two tumor suppressor genes, TSC1 or TSC2, cause tuberous sclerosis complex (TSC), a syndrome resulting in benign hamartomatous tumors and neurological disorders. Cellular growth defects and neuronal disorganization associated with TSC are believed to be due to upregulated TOR signaling. We overexpressed Rheb, an upstream regulator of TOR, in two different subsets of D. melanogaster central brain neurons in order to upregulate the Tsc- Rheb-TOR pathway. Overexpression of Rheb in either the mushroom bodies or the insulin producing cells resulted in enlarged axon projections and cell bodies, which continued to increase in size with prolonged Rheb expression as the animals aged. Additionally, Rheb overexpression in the mushroom bodies resulted in deficiencies in 3 hr but not immediate appetitive memory. Thus, Rheb overexpression in the central brain neurons of flies causes not only morphological phenotypes, but behavioral and aging phenotypes that may mirror symptoms of TSC. Citation: Brown HLD, Kaun KR, Edgar BA (2012) The Small GTPase Rheb Affects Central Brain Neuronal Morphology and Memory Formation in Drosophila. PLoS ONE 7(9): e44888. doi:10.1371/journal.pone.0044888 Editor: Shree Ram Singh, National Cancer Institute, United States of America Received February 28, 2012; Accepted August 14, 2012; Published September 19, 2012 Copyright: ß 2012 Brown et al.