Downloaded from the Same Database
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Abundance of Sesamia Nonagrioides (Lef.) (Lepidoptera: Noctuidae) on the Edges of the Mediterranean Basin
Hindawi Publishing Corporation Psyche Volume 2012, Article ID 854045, 7 pages doi:10.1155/2012/854045 Review Article Abundance of Sesamia nonagrioides (Lef.) (Lepidoptera: Noctuidae) on the Edges of the Mediterranean Basin Matilde Eizaguirre1 and Argyro A. Fantinou2 1 Department of Crop and Forest Sciences, University of Lleida, 25198 Lleida, Spain 2 Laboratory of Ecology, Agricultural University of Athens, 11855 Athens, Greece Correspondence should be addressed to Matilde Eizaguirre, [email protected] Received 5 September 2011; Revised 17 November 2011; Accepted 21 November 2011 Academic Editor: Matilda Savopoulou-Soultani Copyright © 2012 M. Eizaguirre and A. A. Fantinou. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Organisms inhabiting seasonal environments are able to synchronize their life cycles with seasonal cycles of biotic and abiotic factors. Diapause, a state of low metabolic activity and developmental arrest, is used by many insect species to cope with adverse conditions. Sesamia nonagrioides is a serious pest of corn in the Mediterranean regions and Central Africa. It is multivoltine, with two to four generations per year, that overwinters as mature larva in the northern of the Sahara desert. Our purpose was to compare theresponseoftheS. nonagrioides populations occurring in the broader circum-Mediterranean area, with particular attention to the diapause period and the different numbers of generations per season. To this end, we tried to determine whether populations in the area differ in their response to photoperiod and whether we can foresee the number of generations in different areas. -

Walker) (Lepidoptera
Journal of Entomology and Zoology Studies 2018; 6(6): 26-32 E-ISSN: 2320-7078 P-ISSN: 2349-6800 Development of SSRs and its application in JEZS 2018; 6(6): 26-32 © 2018 JEZS genetic diversity study of Indian population of Received: 07-09-2018 Accepted: 09-10-2018 Sesamia inferens (Walker) (Lepidoptera: B Motcha Anthony Reetha Noctuidae) Ph.D. Scholar, Department of Biotechnology, Jain University, Bengaluru, Karnataka, India B Motcha Anthony Reetha and M Mohan M Mohan Principal Scientist, Division of Abstract Germplasm Collection and Pink stem borer (PSB) has become the major pest of cereals in India and other parts of the Asia. The Characterization, ICAR-National wide geographic distribution and broad host range of PSB is likely to result in high genetic variability Bureau of Agricultural Insect Resources, Bangalore, within the species. To understand this in better way we have identified six polymorphic SSRs out of 64 Karnataka, India SSRs developed from 497 genomic DNA sequences available in NCBI database. These six SSRs were able to show the genetic difference among the Sesamia inferens population with respect to their host preference. The result of UPGMA dendrogram and Principal component analysis by using jaccards similarity coefficient data, different populations of S. inferens were clustered according to host. These results suggest a low level of inter-population gene flow in pairwise populations from sorghum, sugarcane, maize and rice fields in India. Such levels of differentiation among populations may indicate only a moderate dispersal capacity of S. inferens, even when no remarkable geographic barriers exist. For an effective management of this pest in the future, there is urgent need for a better understanding of the gene flow of sympatric S. -

The Role of Wild Host Plants in the Abundance of Lepidopteran Stem Borers Along Altitudinal Gradients in Kenya
Ann. soc. enromol. Fr. (n.s.), 2006, 42 (3-4) : 363-370 ARTICLE The role ofwild host plants in the abundance oflepidopteran stem borers along altitudinal gradient in Kenya GEORGE O. ONG'AMO(I), BRUNO P. LE RD(I), STI~,PHANE DUPAS(l), PASCAL MOYAL(l), ERIC MUCHUGU(3), PAUL-ANDRE CALATAYUD(I) & JEAN-FRAN<;:OIS SILVAIN(2) (I) Nocruid Stem Borer Biodiversity Project (NSBB), Insrirur de Recherche pour le Developpernenr I International Cenrre of Insect Physiology and Ecology (IRD/ICIPE), P.O. Box 30772, Nairobi, Kenya (2) IRD, UR R072 clo CNRS, UPR 9034, Laboraroire Evolution, Genomes et Speciarion, avenue de la Terrasse, 91198 Gif/Yvene, France (31 Stem borer Biological Control Project (ICIPE), P.O. Box 30772, Nairobi, Kenya Abstract. Presence of wild host plants of stem borers in cereal-growing areas has been considered as reservoirs of lepidopteran stem borers, responsible for attack of crops during the growing season. Surveys to catalogue hosts and borers as well as to assess the abundance of the hosts were carried out during the cropping and non-cropping seasons in different agro-ecological zones along varying altitude gradient in Kenya. A total of 61 stem borer species belonging to families Noctuidae (25), Crambidae (14), Pyralidae (9), Tortricidae (11) and Cossidae (2) were recovered from 42 wild plant species. Two noctuids, Busseola fusca (Fuller), Sesamia calamistis Hampson, and two crambids, Chilo partellus (Swinhoe) and Chilo orichalcociliellus (Strand) were the four main borer species found associated with maize plants. In the wild, B. fusca was recovered from a limited number of host plant species and among them were Sorghum arundinaceum (Desvaux) Stapf, Setaria megaphylla (Steudel) 1. -

Response of Chilo Partellus (Lepidoptera: Crambidae) to Bt Maize in South Africa
Response of Chilo partellus (Lepidoptera: Crambidae) to Bt maize in South Africa J Vorster orcid.org/0000-0001-8126-6860 Dissertation submitted in fulfilment of the requirements for the Masters degree in Environmental Science at the North-West University Supervisor: Prof J van den Berg Co-supervisor: Prof MJ du Plessis Assistant supervisor: Dr A Erasmus Graduation May 2018 23441674 ACKNOWLEDGEMENTS This dissertation would not have been possible without the help of so many people. I am blessed and very grateful to have them in my life. I would like to start with our God Almighty and our Saviour who bestowed upon me the strength, wisdom and peace of mind to finish this project and who also have sent me these blessed people in my life. I would like to thank Prof. Johnnie van den Berg and Dr. Annemie Erasmus for all the guidance and support they have given me. You taught me that small things can make a big difference. Statistics can be difficult sometimes and I thank Prof. Hannalene du Plessis and Prof. Suria Elis for the help with the statistics. Thank you to all the staff at the ARC-GCI that assisted me with the trials in the lab and the planting. Elrine Strydom, Mabel du Toit, Heidi Meyer and Ursula du Plessis, thank you for the countless after hours we had to spend and for the warm hearted kindness you have given me. I would also like to thank my parents whom I dearly love for all the encouragement and motivation to do my best. You taught me that hard work does not come easily, but the fruit that you pick from it is what motivates us. -

First Detection of a Sesamia Nonagrioides Resistance Allele to Bt Maize in Europe Received: 1 November 2017 Ana M
www.nature.com/scientificreports OPEN First detection of a Sesamia nonagrioides resistance allele to Bt maize in Europe Received: 1 November 2017 Ana M. Camargo1,2, David A. Andow2, Pedro Castañera1 & Gema P. Farinós 1 Accepted: 9 February 2018 The Ebro Valley (Spain) is the only hotspot area in Europe where resistance evolution of target pests Published: xx xx xxxx to Cry1Ab protein is most likely, owing to the high and regular adoption of Bt maize (>60%). The high-dose/refuge (HDR) strategy was implemented to delay resistance evolution, and to be efective it requires the frequency of resistance alleles to be very low (<0.001). An F2 screen was performed in 2016 to estimate the frequency of resistance alleles in Sesamia nonagrioides from this area and to evaluate if the HDR strategy is still working efectively. Out of the 137 isofemale lines screened on Cry1Ab maize leaf tissue, molted larvae and extensive feeding were observed for two consecutive generations in one line, indicating this line carried a resistance allele. The frequency of resistance alleles in 2016 was 0.0036 (CI 95% 0.0004–0.0100), higher but not statistically diferent from the value obtained in 2004– 2005. Resistance does not seem to be evolving faster than predicted by a S. nonagrioides resistance evolution model, but the frequency of resistance is now triple the value recommended for an efective implementation of the HDR strategy. Owing to this, complementary measures should be considered to further delay resistance evolution in the Ebro Valley. Te commercial use of genetically engineered (GE) crops in Europe has been controversial. -

Sesamia Nonagrioides) Under Warming Climate in Europe Andrea Maiorano
Brigham Young University BYU ScholarsArchive 6th International Congress on Environmental International Congress on Environmental Modelling and Software - Leipzig, Germany - July Modelling and Software 2012 Jul 1st, 12:00 AM Potential distribution and phenological development of the Mediterranean Corn Borer (Sesamia nonagrioides) under warming climate in Europe Andrea Maiorano Marcello Donatelli Davide Fumagalli Follow this and additional works at: https://scholarsarchive.byu.edu/iemssconference Maiorano, Andrea; Donatelli, Marcello; and Fumagalli, Davide, "Potential distribution and phenological development of the Mediterranean Corn Borer (Sesamia nonagrioides) under warming climate in Europe" (2012). International Congress on Environmental Modelling and Software. 324. https://scholarsarchive.byu.edu/iemssconference/2012/Stream-B/324 This Event is brought to you for free and open access by the Civil and Environmental Engineering at BYU ScholarsArchive. It has been accepted for inclusion in International Congress on Environmental Modelling and Software by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. International Environmental Modelling and Software Society (iEMSs) 2012 International Congress on Environmental Modelling and Software Managing Resources of a Limited Planet, Sixth Biennial Meeting, Leipzig, Germany R. Seppelt, A.A. Voinov, S. Lange, D. Bankamp (Eds.) http://www.iemss.org/society/index.php/iemss-2012-proceedings Potential distribution and phenological development of the Mediterranean Corn Borer (Sesamia nonagrioides) under warming climate in Europe Andrea Maiorano 1, Marcello Donatelli 1,2, Davide Fumagalli 1 1 European Commission, Joint Research Centre, Institute for Environment and Sustainability, MARS Unit – H04 AGRI4CAST, Ispra, Italy 2 CRA-CIN, Agriculture Research Council, Italy 1 [email protected] Abstract: Climate change has the potential to modify the current distribution and phenological development of insect pests. -

Downloaded from BOLD Or Requested from Other Authors
www.nature.com/scientificreports OPEN Towards a global DNA barcode reference library for quarantine identifcations of lepidopteran Received: 28 November 2018 Accepted: 5 April 2019 stemborers, with an emphasis on Published: xx xx xxxx sugarcane pests Timothy R. C. Lee 1, Stacey J. Anderson2, Lucy T. T. Tran-Nguyen3, Nader Sallam4, Bruno P. Le Ru5,6, Desmond Conlong7,8, Kevin Powell 9, Andrew Ward10 & Andrew Mitchell1 Lepidopteran stemborers are among the most damaging agricultural pests worldwide, able to reduce crop yields by up to 40%. Sugarcane is the world’s most prolifc crop, and several stemborer species from the families Noctuidae, Tortricidae, Crambidae and Pyralidae attack sugarcane. Australia is currently free of the most damaging stemborers, but biosecurity eforts are hampered by the difculty in morphologically distinguishing stemborer species. Here we assess the utility of DNA barcoding in identifying stemborer pest species. We review the current state of the COI barcode sequence library for sugarcane stemborers, assembling a dataset of 1297 sequences from 64 species. Sequences were from specimens collected and identifed in this study, downloaded from BOLD or requested from other authors. We performed species delimitation analyses to assess species diversity and the efectiveness of barcoding in this group. Seven species exhibited <0.03 K2P interspecifc diversity, indicating that diagnostic barcoding will work well in most of the studied taxa. We identifed 24 instances of identifcation errors in the online database, which has hampered unambiguous stemborer identifcation using barcodes. Instances of very high within-species diversity indicate that nuclear markers (e.g. 18S, 28S) and additional morphological data (genitalia dissection of all lineages) are needed to confrm species boundaries. -

The Apameini of Israel (Lepidoptera: Noctuidae) SHILAP Revista De Lepidopterología, Vol
SHILAP Revista de Lepidopterología ISSN: 0300-5267 [email protected] Sociedad Hispano-Luso-Americana de Lepidopterología España Kravchenko, V. D.; Fibiger, M.; Mooser, J.; Junnila, A.; Müller, G. C. The Apameini of Israel (Lepidoptera: Noctuidae) SHILAP Revista de Lepidopterología, vol. 36, núm. 142, junio, 2008, pp. 253-259 Sociedad Hispano-Luso-Americana de Lepidopterología Madrid, España Available in: http://www.redalyc.org/articulo.oa?id=45512540015 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative SHILAP Nº 142 9/6/08 11:52 Página 253 SHILAP Revta. lepid., 36 (142), junio 2008: 253-259 CODEN: SRLPEF ISSN:0300-5267 The Apameini of Israel (Lepidoptera: Noctuidae) V. D. Kravchenko, M. Fibiger, J. Mooser, A. Junnila & G. C. Müller Abstract In Israel, 20 species of tribe Apameini belonging to 10 genera have been found to date. Four species are endemic of the Levant (Sesamia ilonae, Luperina kravchenkoi, Gortyna gyulaii and Lenisa wiltshirei). Others are mostly Palaearctic, Mediterranean, Iranian and Irano-Turanian elements. Grassland species of the Apameini are mainly associated with the Temperate region and are univoltine with highest rate of occurrence in May, or in autumn. Most wetland and oases species are multivoltine and occur in oases and riverbeds over the country, though few of the species are extremely localized. The S. ilonae is presently known only from northern area of the Sea of Galilee, while A. deserticola is from oases of the Dead Sea area. -

Literature Search for Annual Post Market Environmental Monitoring Report of Mon 810 Maize
LITERATURE SEARCH FOR ANNUAL POST MARKET ENVIRONMENTAL MONITORING REPORT OF MON 810 MAIZE Appendix 5. Annual Monitoring Report on the cultivation of MON 810 in the 2018 growing season Bayer Agriculture BVBA, December 2019 1 Table of contents 1. INTRODUCTION............................................................................................................ 3 2. IDENTIFYING THE REVIEW QUESTION AND PURPOSE FOR UNDERTAKING THE LITERATURE SEARCH............................................................... 3 3. SEARCHING FOR IDENTIFYING RELEVANT STUDIES .................................... 4 3.1. SEARCH TERMS AND THEIR COMBINATION .................................................................. 4 3.2. LIMITS APPLIED ........................................................................................................... 6 3.3. LANGUAGE ................................................................................................................. 7 3.4. TIME PERIOD ............................................................................................................... 7 3.5. REFERENCE STUDIES ................................................................................................... 7 3.6. INFORMATION SOURCES ............................................................................................ 13 3.6.1. Electronic bibliographic databases ................................................................. 13 3.6.2. Relevant key organisations ............................................................................. -
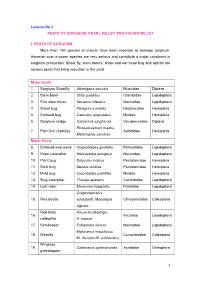
Lecture No 3 PESTS of SORGHUM, PEARL MILLET and FINGER MILLET
Lecture No 3 PESTS OF SORGHUM, PEARL MILLET AND FINGER MILLET I. PESTS OF SORGHUM More than 150 species of insects have been reported to damage sorghum. However over a dozen species are very serious and constitute a major constraint in sorghum production. Shoot fly, stem borers, shoot and ear head bug and aphids are serious pests that bring reduction in the yield. Major pests 1. Sorghum Shootfly Atherigona soccata Muscidae Diptera 2. Stem borer Chilo partellus Crambidae Lepidoptera 3. Pink stem borer Sesamia inferens Noctuidae Lepidoptera 4 Shoot bug Peregrinus maidis Delphacidae Hemiptera 5. Earhead bug Calocoris angustatus Miridae Hemiptera 6. Sorghum midge Contarinia sorghicola Cecidomyiidae Diptera Rhopalosiphum maidis, 7. Plant lice (Aphids) Aphididae Hemiptera Melanaphis sacchari Minor Pests 8. Earhead web worm Cryptoblabes gnidiella Pyraustidae Lepidoptera 9. Gram caterpillar Helicoverpa armigera Noctuidae Lepidoptera 10. Plant bug Dolycoris indicus Pentatomidae Hemiptera 11. Stink bug Nezara viridula Pentatomidae Hemiptera 12. Mirid bug Creontiades pallidifer Miridae Hemiptera 13. Slug caterpillar Thosea apierens Cochlididae Lepidoptera 14. Leaf roller Marasmia trapezalis Pyralidae Lepidoptera Cryptocephalus 15. Flea beetle schestedii, Monolepta Chrysomelidae Coleoptera signata Red hairy Amsacta albistriga, 16. Arctiidae Lepidoptera caterpillar A. moorei 17. Semilooper Eublemma silicula Noctuidae Lepidoptera Myllocerus maculosus 18. Weevils Curculionidae Coleoptera M. discolor,M. subfaciatus Wingless 19. Colemania sphenaroides Acrididae Orthoptera grasshopper MAJOR PESTS 1.Sorghum Shootfly: Atherigona soccata (Muscidae: Diptera) Distribution and status Maharashtra, Andhra Pradesh, Tamil Nadu and Karnataka Host range: Maize, ragi, bajra, rice, wheat and grasses Damage symptoms The maggot on hatching migrates to the upper surface of leaf and enters between the leaf sheath and stem. -

Crambidae Biosecurity Occurrence Background Subfamilies Short Description Diagnosis
Diaphania nitidalis Chilo infuscatellus Crambidae Webworms, Grass Moths, Shoot Borers Biosecurity BIOSECURITY ALERT This Family is of Biosecurity Concern Occurrence This family occurs in Australia. Background The Crambidae is a large, diverse and ubiquitous family of moths that currently comprises 11,500 species globally, with at least half that number again undescribed. The Crambidae and the Pyralidae constitute the superfamily Pyraloidea. Crambid larvae are concealed feeders with a great diversity in feeding habits, shelter building and hosts, such as: leaf rollers, shoot borers, grass borers, leaf webbers, moss feeders, root feeders that shelter in soil tunnels, and solely aquatic life habits. Many species are economically important pests in crops and stored food products. Subfamilies Until recently, the Crambidae was treated as a subfamily under the Pyralidae (snout moths or grass moths). Now they form the superfamily Pyraloidea with the Pyralidae. The Crambidae currently consists of the following 14 subfamilies: Acentropinae Crambinae Cybalomiinae Glaphyriinae Heliothelinae Lathrotelinae Linostinae Midilinae Musotiminae Odontiinae Pyraustinae Schoenobiinae Scopariinae Spilomelinae Short Description Crambid caterpillars are generally cylindrical, with a semiprognathous head and only primary setae (Fig 1). They are often plainly coloured (Fig. 16, Fig. 19), but can be patterned with longitudinal stripes and pinacula that may give them a spotted appearance (Fig. 10, Fig. 11, Fig. 14, Fig. 22). Prolegs may be reduced in borers (Fig. 16). More detailed descriptions are provided below. This factsheet presents, firstly, diagnostic features for the Pyraloidea (Pyralidae and Crambidae) and then the Crambidae. Information and diagnostic features are then provided for crambids listed as priority biosecurity threats for northern Australia. -

Assessment of Grain Yield Losses in Pearl Millet Due to the Millet Stemborer, Coniesta Ignefusalis (Hampson)
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Faculty Publications: Department of Entomology Entomology, Department of 2003 ASSESSMENT OF GRAIN YIELD LOSSES IN PEARL MILLET DUE TO THE MILLET STEMBORER, CONIESTA IGNEFUSALIS (HAMPSON) Aissetou Drame-Yaye Université Abdou Moumouni Ousmane Youm University of Nebraska-Lincoln, [email protected] Jonathan N. Ayertey University of Ghana Follow this and additional works at: https://digitalcommons.unl.edu/entomologyfacpub Part of the Entomology Commons Drame-Yaye, Aissetou; Youm, Ousmane; and Ayertey, Jonathan N., "ASSESSMENT OF GRAIN YIELD LOSSES IN PEARL MILLET DUE TO THE MILLET STEMBORER, CONIESTA IGNEFUSALIS (HAMPSON)" (2003). Faculty Publications: Department of Entomology. 328. https://digitalcommons.unl.edu/entomologyfacpub/328 This Article is brought to you for free and open access by the Entomology, Department of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Faculty Publications: Department of Entomology by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Insect Sci. Applic. Vol. 23, No. 3,Coniesta pp. 259–265, ignefusalis 2003 larval establishment and damage 0191-9040/03 $3.00 + 0.00 259 Printed in Kenya. All rights reserved © 2003 ICIPE ASSESSMENT OF GRAIN YIELD LOSSES IN PEARL MILLET DUE TO THE MILLET STEMBORER, CONIESTA IGNEFUSALIS (HAMPSON) AISSETOU DRAME-YAYE1, OUSMANE YOUM2 AND JONATHAN N. AYERTEY3 1Faculté d’Agronomie, Université Abdou Moumouni , BP 12040 Niamey, Niger; 2 ICRISAT Sahelian Centre, BP 12404, Niamey, Niger; 3 Department of Crop Science, University of Ghana, P. O. Box LG 44, Legon Accra, Ghana (Accepted 4 June 2003) Abstract—Studies were conducted at the ICRISAT Sahelian Centre, Niger, to assess damage and yield loss by the millet stemborer, Coniesta ignefusalis (Lepidoptera: Pyralidae) on Pennisetum glaucum (L.) R.