Metabolomics Analysis of Skeletal Muscles from FKRP-Deficient Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Targeted Metabolic Labeling of Yeast N-Glycans with Unnatural Sugars
Targeted metabolic labeling of yeast N-glycans with unnatural sugars Mark A. Breidenbacha, Jennifer E. G. Gallagherb, David S. Kingc, Brian P. Smarta, Peng Wua,2, and Carolyn R. Bertozzia,b,c,d,1 aDepartments of Chemistry; bMolecular and Cell Biology; and cHoward Hughes Medical Institute, University of California, Berkeley, CA 94720; and dThe Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA 94720 Edited by David A. Tirrell, California Institute of Technology, Pasadena, CA, and approved December 23, 2009 (received for review September 30, 2009) Metabolic labeling of glycans with synthetic sugar analogs has and mass spectrometric analyses of proteins displaying the nonna- emerged as an attractive means for introducing nonnatural chemi- tural chemical functionality (5, 6). cal functionality into glycoproteins. However, the complexities of While existing glycan labeling methodology is suitable for stud- glycan biosynthesis prevent the installation of nonnatural moieties ies that require only stochastic insertion of analogs at low levels, at defined, predictable locations within glycoproteins at high levels the technique is inadequate for applications in which specific of incorporation. Here, we demonstrate that the conserved monosaccharides must be reliably targeted for metabolic replace- N-acetyglucosamine (GlcNAc) residues within chitobiose cores of ment. For example, high-efficiency and predictable installation of N-glycans in the model organism Saccharomyces cerevisiae can sugar analogs could dramatically facilitate biophysical studies of be specifically targeted for metabolic replacement by unnatural glycoprotein structure and function via site-specific introduction sugars. We introduced an exogenous GlcNAc salvage pathway into of fluorophores and heavy atoms. Unfortunately, site-specific me- yeast, allowing cells to metabolize GlcNAc provided as a supple- tabolic labeling of glycans is hindered by multiple factors. -

High Affinity Biomolecular Interactions That Can Mediate Binding of Pathogenic Bacteria to Host Cells
Glycan:glycan interactions: High affinity biomolecular interactions that can mediate binding of pathogenic bacteria to host cells Christopher J. Daya,1, Elizabeth N. Tranb, Evgeny A. Semchenkoa, Greg Trama, Lauren E. Hartley-Tassella, Preston S. K. Nga, Rebecca M. Kinga, Rachel Ulanovskya, Sarah McAtamneya, Michael A. Apicellac, Joe Tiralongoa, Renato Moronab,2, Victoria Korolika,2, and Michael P. Jenningsa,1,2 aInstitute for Glycomics, Griffith University Gold Coast Campus, Gold Coast, QLD 4222, Australia; bSchool of Biological Sciences, Department of Molecular and Cellular Biology, University of Adelaide, Adelaide, SA 5005, Australia; and cDepartment of Microbiology, University of Iowa, Iowa City, IA 52242 Edited by Rino Rappuoli, GSK Vaccines, Siena, Italy, and approved November 10, 2015 (received for review November 3, 2014) Cells from all domains of life express glycan structures attached to Interestingly, there are specific reports of several bacteria lipids and proteins on their surface, called glycoconjugates. Cell-to- expressing truncated surface polysaccharides and oligosaccharides cell contact mediated by glycan:glycan interactions have been that are significantly less adherent than wild-type equivalents considered to be low-affinity interactions that precede high- (10, 11), or that their adherence can be blocked by extracted affinity protein–glycan or protein–protein interactions. In several LOS/LPS (10), indicating a role for bacterial surface glycans in pathogenic bacteria, truncation of surface glycans, lipooligosac- adherence to host cells. This decreased adherence of rough strains charide (LOS), or lipopolysaccharide (LPS) have been reported to or blocking of adherence using the free lipooligosaccharide (LOS)/ significantly reduce bacterial adherence to host cells. Here, we lipopolysaccharide (LPS) in both cell-based and animal infection show that the saccharide component of LOS/LPS have direct, models has been noted in a range of Gram-negative bacteria in- high-affinity interactions with host glycans. -

A Systematic Review on the Implications of O-Linked Glycan Branching and Truncating Enzymes on Cancer Progression and Metastasis
cells Review A Systematic Review on the Implications of O-linked Glycan Branching and Truncating Enzymes on Cancer Progression and Metastasis 1, 1, 1 1,2,3, Rohitesh Gupta y, Frank Leon y, Sanchita Rauth , Surinder K. Batra * and Moorthy P. Ponnusamy 1,2,* 1 Department of Biochemistry and Molecular Biology, University of Nebraska Medical Center, Omaha, NE 68105, USA; [email protected] (R.G.); [email protected] (F.L.); [email protected] (S.R.) 2 Fred and Pamela Buffett Cancer Center, Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE 681980-5900, USA 3 Department of Pathology and Microbiology, UNMC, Omaha, NE 68198-5900, USA * Correspondence: [email protected] (S.K.B.); [email protected] (M.P.P.); Tel.: +402-559-5455 (S.K.B.); +402-559-1170 (M.P.P.); Fax: +402-559-6650 (S.K.B. & M.P.P.) Equal contribution. y Received: 21 January 2020; Accepted: 12 February 2020; Published: 14 February 2020 Abstract: Glycosylation is the most commonly occurring post-translational modifications, and is believed to modify over 50% of all proteins. The process of glycan modification is directed by different glycosyltransferases, depending on the cell in which it is expressed. These small carbohydrate molecules consist of multiple glycan families that facilitate cell–cell interactions, protein interactions, and downstream signaling. An alteration of several types of O-glycan core structures have been implicated in multiple cancers, largely due to differential glycosyltransferase expression or activity. Consequently, aberrant O-linked glycosylation has been extensively demonstrated to affect biological function and protein integrity that directly result in cancer growth and progression of several diseases. -

Biomolecule Purification, Characterization, and Analyses Biomolecule Purification, Characterization, and Analyses Characterization, Purification, Biomolecule
Biomolecule Purification, Characterization, and Analyses Biomolecule Purification, Characterization, Analyses and “Quality is always about having a consistent product.” ~ Aoife Hayes, Technical Service Manager, Wexford, Ireland 239 Benchmarking, Method Development, and Troubleshooting: MassPREP Contents Peptide Standard .......................................................273 Phosphorylated Peptide Standards and Sample Preparation Kits..........................273 Innovative HPLC, UHPLC, Delta-Pak HPLC and UHPLC and UPLC Chemistry Consumables Columns ..............................................................................274 for Bioseparations ....................................................241 RapiGest SF Surfactant for Protein Digestions .....................................................275 Amino Acids ................................................................242 AccQ•Tag Ultra Derivatization Protein Separations ...............................................276 Reaction ..............................................................................242 ACQUITY UPLC SEC System Solution ..276 AccQ•Tag Amino Acid Analysis Turn Key Solution .......................................................243 Waters Insulin HMWP HPLC and UHPLC Columns .............................................278 Glycans and Glycoproteins ..............................247 XBridge Protein BEH SEC, 125Å, HILIC for Released 200Å, and 450Å Columns and N-Glycan Analysis .....................................................248 Protein Standard Test -

Lacto Series Glycolipid Biosynthesis Impairs Α2-6 Sialylation On
www.nature.com/scientificreports OPEN Altered (neo-) lacto series glycolipid biosynthesis impairs α2-6 sialylation on N-glycoproteins in Received: 24 November 2016 Accepted: 15 February 2017 ovarian cancer cells Published: 30 March 2017 Shahidul Alam1,2,*, Merrina Anugraham1,*, Yen-Lin Huang1, Reto S. Kohler1, Timm Hettich3, Katharina Winkelbach1, Yasmin Grether1, Mónica Núñez López1, Nailia Khasbiullina4, Nicolai V. Bovin4, Götz Schlotterbeck3 & Francis Jacob1,2 The (neo-) lacto series glycosphingolipids (nsGSLs) comprise of glycan epitopes that are present as blood group antigens, act as primary receptors for human pathogens and are also increasingly associated with malignant diseases. Beta-1, 3-N-acetyl-glucosaminyl-transferase 5 (B3GNT5) is suggested as the key glycosyltransferase for the biosynthesis of nsGSLs. In this study, we investigated the impact of CRISPR-Cas9 -mediated gene disruption of B3GNT5 (∆B3GNT5) on the expression of glycosphingolipids and N-glycoproteins by utilizing immunostaining and glycomics-based PGC-UHPLC- ESI-QTOF-MS/MS profiling.∆ B3GNT5 cells lost nsGSL expression coinciding with reduction of α2-6 sialylation on N-glycoproteins. In contrast, disruption of B4GALNT1, a glycosyltransferase for ganglio series GSLs did not affectα 2-6 sialylation on N-glycoproteins. We further profiled all knownα 2-6 sialyltransferase-encoding genes and showed that the loss of α2-6 sialylation is due to silencing of ST6GAL1 expression in ∆B3GNT5 cells. These results demonstrate that nsGSLs are part of a complex network affectingN -glycosylation in ovarian cancer cells. Glycosphingolipids (GSLs) have been shown to be essential in a wide variety of biological events - such as cell signalling, modification of insulin and EGF-receptor activities, and modulation of Notch ligand activity in Drosophila1. -

Glycosylation States on Intact Proteins Determined by NMR Spectroscopy
molecules Article Glycosylation States on Intact Proteins Determined by NMR Spectroscopy Audra A. Hargett 1, Aaron M. Marcella 1, Huifeng Yu 1, Chao Li 2 , Jared Orwenyo 2, Marcos D. Battistel 1, Lai-Xi Wang 2 and Darón I. Freedberg 1,* 1 Center for Biologics Evaluation and Review, Laboratory of Bacterial Polysaccharides, Food and Drug Administration (FDA), Silver Spring, MD 20993, USA; [email protected] (A.A.H.); [email protected] (A.M.M.); [email protected] (H.Y.); [email protected] (M.D.B.) 2 Department of Chemistry and Biochemistry, University of Maryland, College Park, MD 20742, USA; [email protected] (C.L.); [email protected] (J.O.); [email protected] (L.-X.W.) * Correspondence: [email protected] Abstract: Protein glycosylation is important in many organisms for proper protein folding, signaling, cell adhesion, protein-protein interactions, and immune responses. Thus, effectively determining the extent of glycosylation in glycoprotein therapeutics is crucial. Up to now, characterizing protein glycosylation has been carried out mostly by liquid chromatography mass spectrometry (LC-MS), which requires careful sample processing, e.g., glycan removal or protein digestion and glycopeptide enrichment. Herein, we introduce an NMR-based method to better characterize intact glycoproteins in natural abundance. This non-destructive method relies on exploiting differences in nuclear relaxation to suppress the NMR signals of the protein while maintaining glycan signals. Using RNase B Man5 and RNase B Man9, we establish reference spectra that can be used to determine the different Citation: Hargett, A.A.; Marcella, glycoforms present in heterogeneously glycosylated commercial RNase B. -

Interpretive Guide
INTERPRETIVE GUIDE Contents INTRODUCTION .........................................................................1 NUTREVAL BIOMARKERS ...........................................................5 Metabolic Analysis Markers ....................................................5 Malabsorption and Dysbiosis Markers .....................................5 Cellular Energy & Mitochondrial Metabolites ..........................6 Neurotransmitter Metabolites ...............................................8 Vitamin Markers ....................................................................9 Toxin & Detoxification Markers ..............................................9 Amino Acids ..........................................................................10 Essential and Metabolic Fatty Acids .........................................13 Cardiovascular Risk ................................................................15 Oxidative Stress Markers ........................................................16 Elemental Markers ................................................................17 Toxic Elements .......................................................................18 INTERPRETATION-AT-A-GLANCE .................................................19 REFERENCES .............................................................................23 INTRODUCTION A shortage of any nutrient can lead to biochemical NutrEval profile evaluates several important biochemical disturbances that affect healthy cellular and tissue pathways to help determine nutrient -

Enzymatic Passaging of Human Embryonic Stem Cells Alters Central Carbon Metabolism and Glycan Abundance
UC San Diego UC San Diego Previously Published Works Title Enzymatic passaging of human embryonic stem cells alters central carbon metabolism and glycan abundance Permalink https://escholarship.org/uc/item/1x27t1sx Journal Biotechnology Journal, 10(10) ISSN 1860-6768 Authors Badur, MG Zhang, H Metallo, CM Publication Date 2015-10-01 DOI 10.1002/biot.201400749 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Biotechnology Biotechnol. J. 2015, 10 DOI 10.1002/biot.201400749 Journal www.biotechnology-journal.com Research Article Enzymatic passaging of human embryonic stem cells alters central carbon metabolism and glycan abundance Mehmet G. Badur1, Hui Zhang1 and Christian M. Metallo1,2 1 Department of Bioengineering, University of California, San Diego, La Jolla, USA 2 Moores Cancer Center, University of California, San Diego, La Jolla, USA To realize the potential of human embryonic stem cells (hESCs) in regenerative medicine and drug Received 17 MAR 2015 discovery applications, large numbers of cells that accurately recapitulate cell and tissue function Revised 10 JUL 2015 must be robustly produced. Previous studies have suggested that genetic instability and epigenetic Accepted 18 AUG 2015 changes occur as a consequence of enzymatic passaging. However, the potential impacts of such Accepted passaging methods on the metabolism of hESCs have not been described. Using stable isotope article online 18 AUG 2015 tracing and mass spectrometry-based metabolomics, we have explored how different passaging reagents impact hESC metabolism. Enzymatic passaging caused significant decreases in glucose utilization throughout central carbon metabolism along with attenuated de novo lipogenesis. In addition, we developed and validated a method for rapidly quantifying glycan abundance and isotopic labeling in hydrolyzed biomass. -

Important Roles of Dietary Taurine, Creatine, Carnosine, Anserine and 4‑Hydroxyproline in Human Nutrition and Health
Amino Acids (2020) 52:329–360 https://doi.org/10.1007/s00726-020-02823-6 INVITED REVIEW Important roles of dietary taurine, creatine, carnosine, anserine and 4‑hydroxyproline in human nutrition and health Guoyao Wu1 Received: 25 October 2019 / Accepted: 29 January 2020 / Published online: 18 February 2020 © The Author(s) 2020 Abstract Taurine (a sulfur-containing β-amino acid), creatine (a metabolite of arginine, glycine and methionine), carnosine (a dipep- tide; β-alanyl-L-histidine), and 4-hydroxyproline (an imino acid; also often referred to as an amino acid) were discovered in cattle, and the discovery of anserine (a methylated product of carnosine; β-alanyl-1-methyl-L-histidine) also originated with cattle. These fve nutrients are highly abundant in beef, and have important physiological roles in anti-oxidative and anti- infammatory reactions, as well as neurological, muscular, retinal, immunological and cardiovascular function. Of particular note, taurine, carnosine, anserine, and creatine are absent from plants, and hydroxyproline is negligible in many plant-source foods. Consumption of 30 g dry beef can fully meet daily physiological needs of the healthy 70-kg adult human for taurine and carnosine, and can also provide large amounts of creatine, anserine and 4-hydroxyproline to improve human nutrition and health, including metabolic, retinal, immunological, muscular, cartilage, neurological, and cardiovascular health. The present review provides the public with the much-needed knowledge of nutritionally and physiologically signifcant amino acids, dipeptides and creatine in animal-source foods (including beef). Dietary taurine, creatine, carnosine, anserine and 4-hydroxyproline are benefcial for preventing and treating obesity, cardiovascular dysfunction, and ageing-related disorders, as well as inhibiting tumorigenesis, improving skin and bone health, ameliorating neurological abnormalities, and promoting well being in infants, children and adults. -

A Site-Specific Map of the Human Plasma Glycome and Its Age And
www.nature.com/scientificreports OPEN A site‑specifc map of the human plasma glycome and its age and gender‑associated alterations Alexander A. Merleev1, Dayoung Park2,3, Yixuan Xie3, Muchena J. Kailemia3, Gege Xu3, L. Renee Ruhaak3,4, Kyoungmi Kim5, Qiuting Hong3, Qiongyu Li3, Forum Patel1, Yu‑Jui Yvonne Wan6, Alina I. Marusina1, Iannis E. Adamopoulos7,8, Nelvish N. Lal1, Anupum Mitra5, Stephanie T. Le1, Michiko Shimoda1, Guillaume Luxardi1, Carlito B. Lebrilla3,9,10* & Emanual Maverakis1* Alterations in the human glycome have been associated with cancer and autoimmunity. Thus, constructing a site‑specifc map of the human glycome for biomarker research and discovery has been a highly sought‑after objective. However, due to analytical barriers, comprehensive site‑specifc glycoprofling is difcult to perform. To develop a platform to detect easily quantifable, site‑specifc, disease‑associated glycan alterations for clinical applications, we have adapted the multiple reaction monitoring mass spectrometry method for use in glycan biomarker research. The adaptations allow for highly precise site‑specifc glycan monitoring with minimum sample prep. Using this technique, we successfully mapped out the relative abundances of the most common 159 glycopeptides in the plasma of 97 healthy volunteers. This plasma glycome map revealed 796 signifcant (FDR < 0.05) site‑ specifc inter‑protein and intra‑protein glycan associations, of which the vast majority were previously unknown. Since age and gender are relevant covariants in biomarker research, these variables were also characterized. 13 glycopeptides were found to be associated with gender and 41 to be associated with age. Using just fve age‑associated glycopeptides, a highly accurate age prediction model was constructed and validated (r2 = 0.62 ± 0.12). -
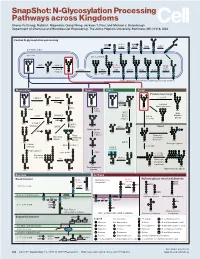
N-Glycosylation Processing Pathways Across Kingdoms Cheng-Yu Chung, Natalia I
SnapShot: N-Glycosylation Processing Pathways across Kingdoms Cheng-Yu Chung, Natalia I. Majewska, Qiong Wang, Jackson T. Paul, and Michael J. Betenbaugh Department of Chemical and Biomolecular Engineering, The Johns Hopkins University, Baltimore, MD 21218, USA Central N-glycosylation processing UDP ALG13 GDP ALG7 ALG14 ALG1 ALG2 ALG11 P P P P P CYTOPLASM P P P P P P GOLGI ER LUMEN GNTI α -Man IA En Bloc ALG3 Transfer ALG9 α-Man IB ER Man I α -Glc I OST ALG10 ALG8 ALG6 ALG12 Asn α -Man IC Asn Asn Asn α -Glc II P P P P P Asn P P P P P ALGs: asparagine-linked N-glycosylation processing enzymes Mammalian Insect Plant Fungi Filamentous fungi α-Man II FUT8 α-1,2-MT XYLT GDP Terminal Asn Asn Asn Asn UDP Asn Galactofuranose GNTII Asn Asn FUT8 f ± α-Gal Asn FUTC3 GNTII High FUT11 α -GALT B14GALTI Och1p Mannose FUT12 glycan α-Man II α-1,2-MT UDP Asn Asn ST3GAL4 Asn Asn GNTIV ST6GAL1 GNTIII GNTV Asn GNase Yeast Hybrid glycan P CMP GNase Asn Asn Tetra- α-Man II Mannosyl- Bisecting antennary phosphate glycan glycan transferase Asn Asn Asn Asn GALT1 Asn ST8SIA2 Core type glycan GNTs B14GALTI α-1,6-MT ST8SIA4 Asn Lewis A structure Polysialylation α-Man PolyLacNAc α-1,2-MT Alternative α-1,3-MT P sialic acid FUT13 ] [ STs Mannosyl- n phosphate Asn Asn Asn transferase Asn Asn Paucimannose Hypermannose glycan Asn Asn Asn glycans Bacteria Archaea Archaea glycan structural diversity Block transfer En Bloc Methanococcus En Bloc Transfer Transfer maripaludis OCH3 Thr OCH AgIB Thr OCH3 3 PERIPLASM PglB B B Asn P - P Asn OSO3 P Asn Asn INNER MEMBRANE P Asn P -

Glycosylation Biomarkers Associated with Age-Related Diseases and Current Methods for Glycan Analysis
International Journal of Molecular Sciences Review Glycosylation Biomarkers Associated with Age-Related Diseases and Current Methods for Glycan Analysis Beatrix Paton 1, Manuel Suarez 2,* , Pol Herrero 1,* and Núria Canela 1 1 Eurecat, Centre Tecnològic de Catalunya, Centre for Omic Sciences, Joint Unit Eurecat-Universitat Rovira i Virgili, Unique Scientific and Technical Infrastructure (ICTS), 43204 Reus, Spain; [email protected] (B.P.); [email protected] (N.C.) 2 Nutrigenomics Research Group, Departament de Bioquímica i Biotecnologia, Universitat Rovira i Virgili, 43007 Tarragona, Spain * Correspondence: [email protected] (M.S.); [email protected] (P.H.) Abstract: Ageing is a complex process which implies the accumulation of molecular, cellular and organ damage, leading to an increased vulnerability to disease. In Western societies, the increase in the elderly population, which is accompanied by ageing-associated pathologies such as cardiovascular and mental diseases, is becoming an increasing economic and social burden for governments. In order to prevent, treat and determine which subjects are more likely to develop these age-related diseases, predictive biomarkers are required. In this sense, some studies suggest that glycans have a potential role as disease biomarkers, as they modify the functions of proteins and take part in intra- and intercellular biological processes. As the glycome reflects the real-time status of these interactions, its characterisation can provide potential diagnostic and prognostic biomarkers for multifactorial diseases. This review gathers the alterations in protein glycosylation profiles that are Citation: Paton, B.; Suarez, M.; Herrero, P.; Canela, N. Glycosylation associated with ageing and age-related diseases, such as cancer, type 2 diabetes mellitus, metabolic Biomarkers Associated with syndrome and several chronic inflammatory diseases.