Deconvolution of Transcriptional Networks Identifies TCF4 As a Master Regulator in Schizophrenia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Computational Analysis of DNA Methylation and Gene Expression Patterns in Prostate Cancer
Ieva Rauluševičiūtė Computational Analysis of DNA Methylation and Gene Expression Patterns in Prostate Cancer Master’s thesis in Molecular Medicine Trondheim, June 2018 Supervisors: dr. Morten Beck Rye and prof. Finn Drabløs Norwegian University of Science and Technology Faculty of Medicine and Health Sciences Department of Clinical and Molecular Medicine ABSTRACT DNA methylation is an important contributor for prostate cancer development and progression. It has been studied experimentally for years, but, lately, high-throughput technologies are able to produce genome-wide DNA methylation data that can be analyzed using various computational approaches. Thus, this study aims to bioinformatically investigate different DNA methylation and gene expression patterns in prostate cancer. DNA methylation data from three datasets (TCGA, Absher and Kirby) was correlated with gene expression data in order to distinguish different regulation patterns. Classically, increased DNA methylation in promoter regions is being associated with gene expression downregulation. Although, results of the present project demonstrate another robust pattern, where DNA hypermethylation in promoter regions of 1,058 common genes is accompanied by upregulated expression. After analyzing expression and methylation values in the same samples from TCGA dataset, expression overcompensation in a dataset as an explanation for upregulation was excluded. Further reasons behind the pattern were investigated using TCGA DNA methylation data with extended list of probes and includes the presence of methylated positions in CpG islands, distance to transcription start sites and alternative TSSs. As compared with the downregulated genes in the classical pattern, upregulated genes were shown to have more positions in CpG islands and closer to TSSs. Moreover, the presence of alternative TSS in prostate was demonstrated, also disclosing the limitations of methylation detection systems. -

The Function and Evolution of C2H2 Zinc Finger Proteins and Transposons
The function and evolution of C2H2 zinc finger proteins and transposons by Laura Francesca Campitelli A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Molecular Genetics University of Toronto © Copyright by Laura Francesca Campitelli 2020 The function and evolution of C2H2 zinc finger proteins and transposons Laura Francesca Campitelli Doctor of Philosophy Department of Molecular Genetics University of Toronto 2020 Abstract Transcription factors (TFs) confer specificity to transcriptional regulation by binding specific DNA sequences and ultimately affecting the ability of RNA polymerase to transcribe a locus. The C2H2 zinc finger proteins (C2H2 ZFPs) are a TF class with the unique ability to diversify their DNA-binding specificities in a short evolutionary time. C2H2 ZFPs comprise the largest class of TFs in Mammalian genomes, including nearly half of all Human TFs (747/1,639). Positive selection on the DNA-binding specificities of C2H2 ZFPs is explained by an evolutionary arms race with endogenous retroelements (EREs; copy-and-paste transposable elements), where the C2H2 ZFPs containing a KRAB repressor domain (KZFPs; 344/747 Human C2H2 ZFPs) are thought to diversify to bind new EREs and repress deleterious transposition events. However, evidence of the gain and loss of KZFP binding sites on the ERE sequence is sparse due to poor resolution of ERE sequence evolution, despite the recent publication of binding preferences for 242/344 Human KZFPs. The goal of my doctoral work has been to characterize the Human C2H2 ZFPs, with specific interest in their evolutionary history, functional diversity, and coevolution with LINE EREs. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

Host Cell Factors Necessary for Influenza a Infection: Meta-Analysis of Genome Wide Studies
Host Cell Factors Necessary for Influenza A Infection: Meta-Analysis of Genome Wide Studies Juliana S. Capitanio and Richard W. Wozniak Department of Cell Biology, Faculty of Medicine and Dentistry, University of Alberta Abstract: The Influenza A virus belongs to the Orthomyxoviridae family. Influenza virus infection occurs yearly in all countries of the world. It usually kills between 250,000 and 500,000 people and causes severe illness in millions more. Over the last century alone we have seen 3 global influenza pandemics. The great human and financial cost of this disease has made it the second most studied virus today, behind HIV. Recently, several genome-wide RNA interference studies have focused on identifying host molecules that participate in Influen- za infection. We used nine of these studies for this meta-analysis. Even though the overlap among genes identified in multiple screens was small, network analysis indicates that similar protein complexes and biological functions of the host were present. As a result, several host gene complexes important for the Influenza virus life cycle were identified. The biological function and the relevance of each identified protein complex in the Influenza virus life cycle is further detailed in this paper. Background and PA bound to the viral genome via nucleoprotein (NP). The viral core is enveloped by a lipid membrane derived from Influenza virus the host cell. The viral protein M1 underlies the membrane and anchors NEP/NS2. Hemagglutinin (HA), neuraminidase Viruses are the simplest life form on earth. They parasite host (NA), and M2 proteins are inserted into the envelope, facing organisms and subvert the host cellular machinery for differ- the viral exterior. -

Comparative Transcriptome Analysis Reveals Key Epigenetic Targets in SARS-Cov-2 Infection
www.nature.com/npjsba ARTICLE OPEN Comparative transcriptome analysis reveals key epigenetic targets in SARS-CoV-2 infection Marisol Salgado-Albarrán 1,2,7, Erick I. Navarro-Delgado 3,7, Aylin Del Moral-Morales 1,7, Nicolas Alcaraz 4, Jan Baumbach 5,6, ✉ ✉ Rodrigo González-Barrios3 and Ernesto Soto-Reyes 1 COVID-19 is an infection caused by SARS-CoV-2 (Severe Acute Respiratory Syndrome coronavirus 2), which has caused a global outbreak. Current research efforts are focused on the understanding of the molecular mechanisms involved in SARS-CoV-2 infection in order to propose drug-based therapeutic options. Transcriptional changes due to epigenetic regulation are key host cell responses to viral infection and have been studied in SARS-CoV and MERS-CoV; however, such changes are not fully described for SARS-CoV-2. In this study, we analyzed multiple transcriptomes obtained from cell lines infected with MERS-CoV, SARS-CoV, and SARS-CoV-2, and from COVID-19 patient-derived samples. Using integrative analyses of gene co-expression networks and de-novo pathway enrichment, we characterize different gene modules and protein pathways enriched with Transcription Factors or Epifactors relevant for SARS-CoV-2 infection. We identified EP300, MOV10, RELA, and TRIM25 as top candidates, and more than 60 additional proteins involved in the epigenetic response during viral infection that has therapeutic potential. Our results show that targeting the epigenetic machinery could be a feasible alternative to treat COVID-19. npj Systems Biology and Applications (2021) 7:21 ; https://doi.org/10.1038/s41540-021-00181-x 1234567890():,; INTRODUCTION hallmark of active chromatin)10, histone acetylation in H3 and The coronavirus family (CoV) are non-segmented, positive-sense, H4 histones, and increased levels of H4K20me2 and unmodified 11 and enveloped RNA viruses that have been identified as the cause H3K36 and H4K79 have been reported . -

Evolutionary Analysis of Viral Sequences in Eukaryotic Genomes
Evolutionary analysis of viral sequences in eukaryotic genomes Sean Schneider A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2014 Reading Committee: James H. Thomas, Chair Willie Swanson Phil Green Program Authorized to Offer Degree: Genome Sciences ©Copyright 2014 Sean Schneider University of Washington Abstract Evolutionary analysis of viral sequences in eukaryotic genomes Sean Schneider Chair of the supervisory committee: Professor James H. Thomas Genome Sciences The focus of this work is several evolutionary analyses of endogenous viral sequences in eukaryotic genomes. Endogenous viral sequences can provide key insights into the past forms and evolutionary history of viruses, as well as the responses of host organisms they infect. In this work I have examined viral sequences in a diverse assortment of eukaryotic hosts in order to study coevolution between hosts and the organisms that infect them. This research consisted of two major lines of investigation. In the first portion of this work, I outline the hypothesis that the C2H2 zinc finger gene family in vertebrates has evolved by birth-death evolution in response to sporadic retroviral infection. The hypothesis suggests an evolutionary model in which newly duplicated zinc finger genes are retained by selection in response to retroviral infection. This hypothesis is supported by a strong association (R2=0.67) between the number of endogenous retroviruses and the number of zinc fingers in diverse vertebrate genomes. Based on this and other evidence, the zinc finger gene family appears to act as a “genomic immune system” against retroviral infections. The other major line of investigation in this work examines endogenous virus sequences utilized by parasitic wasps to disable hosts that they infect. -

(12) United States Patent (10) Patent No.: US 9,605.243 B2 D'amour Et Al
USOO9605243B2 (12) United States Patent (10) Patent No.: US 9,605.243 B2 D'Amour et al. (45) Date of Patent: Mar. 28, 2017 (54) MARKERS OF DEFINITIVE ENDODERM 6,117,986 A 9, 2000 Nardone et al. 6,165,993 A 12/2000 Herrmann et al. (71) Applicant: ViaCyte,7 Inc., San Diego, CA (US) 6,251,6716,200,806 B1 3/20016/2001 ThomsonHogan et al. (72) Inventors: Kevin Allen D'Amour, San Diego, CA 3. R 1658 R.S.A. (US); Alan D. Agulnick, San Diego, 6,506,574 B1 1/2003 Rambhatla et al. CA (US); Emmanuel Edward Baetge, 6,872,389 B1 3/2005 Faris Encinitas, CA (US) 6,921,811 B2 7/2005 Zamora et al. s 7,033,831 B2 4/2006 Fisk et al. 7,153,684 B1 12/2006 Hogan (73) Assignee: ViaCyte, Inc., San Diego, CA (US) 7.256,042 B2 8/2007 Rambhatla et al. - 7,326,572 B2 2/2008 Fisk et al. (*) Notice: Subject to any disclaimer, the term of this 7,510,876 B2 3/2009 D'Amour et al. patent is extended or adjusted under 35 23: R: 258 Nation et al U.S.C. 154(b) by 0 days. 8,586,357- I k B2 11/2013 D'Amour'Amour et al. 2002/00721 17 A1 6/2002 Xu et al. (21) Appl. No.: 14/066,441 2002/0090723 A1 7/2002 Carpenter et al. 2003. O138948 A1 7/2003 Fisk et al. (22) Filed: Oct. 29, 2013 2003. O138949 A1 7/2003 Bhushan et al. -

Transdifferentiation of Human Mesenchymal Stem Cells
Transdifferentiation of Human Mesenchymal Stem Cells Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Julius-Maximilians-Universität Würzburg vorgelegt von Tatjana Schilling aus San Miguel de Tucuman, Argentinien Würzburg, 2007 Eingereicht am: Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Martin J. Müller Gutachter: PD Dr. Norbert Schütze Gutachter: Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am: Hiermit erkläre ich ehrenwörtlich, dass ich die vorliegende Dissertation selbstständig angefertigt und keine anderen als die von mir angegebenen Hilfsmittel und Quellen verwendet habe. Des Weiteren erkläre ich, dass diese Arbeit weder in gleicher noch in ähnlicher Form in einem Prüfungsverfahren vorgelegen hat und ich noch keinen Promotionsversuch unternommen habe. Gerbrunn, 4. Mai 2007 Tatjana Schilling Table of contents i Table of contents 1 Summary ........................................................................................................................ 1 1.1 Summary.................................................................................................................... 1 1.2 Zusammenfassung..................................................................................................... 2 2 Introduction.................................................................................................................... 4 2.1 Osteoporosis and the fatty degeneration of the bone marrow..................................... 4 2.2 Adipose and bone -

Investigating Host Genes Involved In. HIY Control by a Novel Computational Method to Combine GWAS with Eqtl
Investigating Host Genes Involved in. HIY Control by a Novel Computational Method to Combine GWAS with eQTL by Yi Song THESIS Submitted In partial satisfaction of me teqoitements for the degree of MASTER OF SCIENCE In Biological and Medical Informatics In the GRADUATE DIVISION Copyright (2012) by Yi Song ii Acknowledgement First and foremost, I would like to thank my advisor Professor Hao Li, without whom this thesis would not have been possible. I am very grateful that Professor Li lead me into the field of human genomics and gave me the opportunity to pursue this interesting study in his laboratory. Besides the wealth of knowledge and invaluable insights that he offered in every meeting we had, Professor Li is one of the most approachable faculties I have met. I truly appreciate his patient guidance and his enthusiastic supervision throughout my master’s career. I am sincerely thankful to Professor Patricia Babbitt, the Associate Director of the Biomedical Informatics program at UCSF. Over my two years at UCSF, she has always been there to offer her help when I was faced with difficulties. I would also like to thank both Professor Babbitt and Professor Nevan Krogan for investing their valuable time in evaluating my work. I take immense pleasure in thanking my co-workers Dr. Xin He and Christopher Fuller. It has been a true enjoyment to discuss science with Dr. He, whose enthusiasm is a great inspiration to me. I also appreciate his careful editing of my thesis. Christopher Fuller, a PhD candidate in the Biomedical Informatics program, has provided great help for me on technical problems. -

The Changing Chromatome As a Driver of Disease: a Panoramic View from Different Methodologies
The changing chromatome as a driver of disease: A panoramic view from different methodologies Isabel Espejo1, Luciano Di Croce,1,2,3 and Sergi Aranda1 1. Centre for Genomic Regulation (CRG), Barcelona Institute of Science and Technology, Dr. Aiguader 88, Barcelona 08003, Spain 2. Universitat Pompeu Fabra (UPF), Barcelona, Spain 3. ICREA, Pg. Lluis Companys 23, Barcelona 08010, Spain *Corresponding authors: Luciano Di Croce ([email protected]) Sergi Aranda ([email protected]) 1 GRAPHICAL ABSTRACT Chromatin-bound proteins regulate gene expression, replicate and repair DNA, and transmit epigenetic information. Several human diseases are highly influenced by alterations in the chromatin- bound proteome. Thus, biochemical approaches for the systematic characterization of the chromatome could contribute to identifying new regulators of cellular functionality, including those that are relevant to human disorders. 2 SUMMARY Chromatin-bound proteins underlie several fundamental cellular functions, such as control of gene expression and the faithful transmission of genetic and epigenetic information. Components of the chromatin proteome (the “chromatome”) are essential in human life, and mutations in chromatin-bound proteins are frequently drivers of human diseases, such as cancer. Proteomic characterization of chromatin and de novo identification of chromatin interactors could thus reveal important and perhaps unexpected players implicated in human physiology and disease. Recently, intensive research efforts have focused on developing strategies to characterize the chromatome composition. In this review, we provide an overview of the dynamic composition of the chromatome, highlight the importance of its alterations as a driving force in human disease (and particularly in cancer), and discuss the different approaches to systematically characterize the chromatin-bound proteome in a global manner. -
Communication in Asthmatic Airways Inflammation and Intercellular Gene
Downloaded from http://www.jimmunol.org/ by guest on October 2, 2021 is online at: average * The Journal of Immunology , 10 of which you can access for free at: 2011; 186:1861-1869; Prepublished online 27 from submission to initial decision 4 weeks from acceptance to publication December 2010; doi: 10.4049/jimmunol.1002568 http://www.jimmunol.org/content/186/3/1861 Gene Expression Patterns of Th2 Inflammation and Intercellular Communication in Asthmatic Airways David F. Choy, Barmak Modrek, Alexander R. Abbas, Sarah Kummerfeld, Hilary F. Clark, Lawren C. Wu, Grazyna Fedorowicz, Zora Modrusan, John V. Fahy, Prescott G. Woodruff and Joseph R. Arron J Immunol cites 63 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2010/12/27/jimmunol.100256 8.DC1 This article http://www.jimmunol.org/content/186/3/1861.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2011 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of October 2, 2021. The Journal of Immunology Gene Expression Patterns of Th2 Inflammation and Intercellular Communication in Asthmatic Airways David F. -
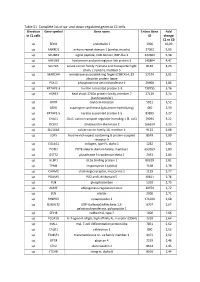
Table S1. Complete List of Up- and Down-Regulated Genes in C1 Cells
Table S1. Complete list of up- and down-regulated genes in C1 cells. Direction Gene symbol Gene name Entrez Gene Fold in C1 cells ID change C1 vs C0 up EDN1 endothelin 1 1906 10,09 up ANKRD1 ankyrin repeat domain 1 (cardiac muscle) 27063 5,95 up SCUBE3 signal peptide, CUB domain, EGF-like 3 222663 5,38 up HAPLN3 hyaluronan and proteoglycan link protein 3 145864 4,47 up SLC7A5 solute carrier family 7 (amino acid transporter light 8140 4,23 chain, L system), member 5 up MARCH4 membrane-associated ring finger (C3HC4) 4, E3 57574 3,91 ubiquitin protein ligase up PSAT1 phosphoserine aminotransferase 1 29968 3,86 up KRTAP2-3 keratin associated protein 2-3 730755 3,76 up HSPB7 heat shock 27kDa protein family, member 7 27129 3,74 (cardiovascular) up OXTR oxytocin receptor 5021 3,52 up ASNS asparagine synthetase (glutamine-hydrolyzing) 440 3,39 up KRTAP1-5 keratin associated protein 1-5 83895 3,27 up CHAC1 ChaC, cation transport regulator homolog 1 (E, coli) 79094 3,22 up DCLK2 doublecortin-like kinase 2 166614 3,15 up SLC16A4 solute carrier family 16, member 4 9122 3,08 up LGR5 leucine-rich repeat containing G protein-coupled 8549 2,99 receptor 5 up COL4A1 collagen, type IV, alpha 1 1282 2,94 up POTEI POTE ankyrin domain family, member I 653269 2,89 up GSTT2 glutathione S-transferase theta 2 2953 2,84 up ULBP1 UL16 binding protein 1 80329 2,81 up TPM1 tropomyosin 1 (alpha) 7168 2,78 up CHRM2 cholinergic receptor, muscarinic 2 1129 2,77 up PDLIM5 PDZ and LIM domain 5 10611 2,76 up PLN phospholamban 5350 2,75 up ADIRF adipogenesis regulatory factor