Small-Molecule Cavα1⋅Cavβ Antagonist Suppresses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Redalyc.Neurobiological Alterations in Alcohol Addiction: a Review
Adicciones ISSN: 0214-4840 [email protected] Sociedad Científica Española de Estudios sobre el Alcohol, el Alcoholismo y las otras Toxicomanías España Erdozain, Amaia M.; Callado, Luis F. Neurobiological alterations in alcohol addiction: a review Adicciones, vol. 26, núm. 4, octubre-diciembre, 2014, pp. 360-370 Sociedad Científica Española de Estudios sobre el Alcohol, el Alcoholismo y las otras Toxicomanías Palma de Mallorca, España Available in: http://www.redalyc.org/articulo.oa?id=289132934009 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative revisión adicciones vol. 26, nº 3 · 2014 Neurobiological alterations in alcohol addiction: a review Alteraciones neurobiológicas en el alcoholismo: revisión Amaia M. Erdozain*,*** and Luis F. Callado*,** *Department of Pharmacology, University of the Basque Country UPV/EHU, Leioa, Bizkaia, Spain and Centro de Investigación Biomédica en Red de Salud Mental (CIBERSAM), Spain. **Biocruces Health Research Institute, Bizkaia, Spain. ***Neuroscience Paris Seine, Université Pierre et Marie Curie, Paris, France Resumen Abstract Todavía se desconoce el mecanismo exacto mediante el cual el etanol The exact mechanism by which ethanol exerts its effects on the brain produce sus efectos en el cerebro. Sin embargo, hoy en día se sabe is still unknown. However, nowadays it is well known that ethanol que el etanol interactúa con proteínas específicas de la membrana interacts with specific neuronal membrane proteins involved in neuronal, implicadas en la transmisión de señales, produciendo así signal transmission, resulting in changes in neural activity. -

Mechanism-Specific Assay Design Facilitates the Discovery of Nav1.7
Mechanism-specific assay design facilitates the PNAS PLUS discovery of Nav1.7-selective inhibitors Tania Chernov-Rogana, Tianbo Lia, Gang Lua, Henry Verschoofb, Kuldip Khakhb, Steven W. Jonesa, Maureen H. Beresinia, Chang Liua, Daniel F. Ortwinec, Steven J. McKerrallc, David H. Hackosd, Daniel Sutherlinc, Charles J. Cohenb, and Jun Chena,1 aDepartment of Biochemical and Cellular Pharmacology, Genentech Inc., San Francisco, CA 94080; bXenon Pharmaceuticals, Burnaby, BC V5G 4W8, Canada; cDepartment of Chemistry, Genentech Inc., San Francisco, CA 94080; and dDepartment of Neuroscience, Genentech Inc., San Francisco, CA 94080 Edited by Bruce P. Bean, Harvard Medical School, Boston, MA, and approved December 11, 2017 (received for review August 30, 2017) Many ion channels, including Nav1.7, Cav1.3, and Kv1.3, are linked therefore are nonselective (14). Recently, a group of arylsulfo- to human pathologies and are important therapeutic targets. To namides was identified as selective Nav1.7 inhibitors (15, 16). develop efficacious and safe drugs, subtype-selective modulation These compounds were discovered empirically, before our cur- is essential, but has been extremely difficult to achieve. We rent understanding that they bind to the voltage-sensing domain postulate that this challenge is caused by the poor assay design, 4 (VSD4) instead of the central cavity. However, PF-771, the and investigate the Nav1.7 membrane potential assay, one of the most advanced compound, was recently halted from clinical most extensively employed screening assays in modern drug development, raising concerns about this chemical class. discovery. The assay uses veratridine to activate channels, and To identify subtype-selective chemical scaffolds, large libraries compounds are identified based on the inhibition of veratridine- of compounds, sometimes exceeding millions of individual evoked activities. -

Is Diabetic Nerve Pain Caused by Dysregulated Ion Channels in Sensory Neurons?
Diabetes Volume 64, December 2015 3987 Slobodan M. Todorovic Is Diabetic Nerve Pain Caused by Dysregulated Ion Channels in Sensory Neurons? Diabetes 2015;64:3987–3989 | DOI: 10.2337/dbi15-0006 In diabetes, a common and debilitating chronic disease, nociceptive sensory neurons, also known as dorsal root peripheral diabetic neuropathy (PDN) is the most frequent ganglion (DRG) neurons, which play a critical role in complication, occurring in about two-thirds of the patients modulating overall cellular excitability. These findings are (1,2). At least one-third of patients with diabetes experience both important and relevant, as increased excitability of painful symptoms including hyperalgesia and/or allodynia as sensory neurons is believed to contribute directly to the well as spontaneous pain in the form of burning or tingling, development and maintenance of painful symptoms, in- despite the degeneration of peripheral nerves (3). Eventually, cluding hyperalgesia, allodynia, and/or spontaneous pain. these painful symptoms usually subside as the disabling pain Recent studies have shown that the CaV3.2 isoform of is replaced by the complete loss of sensation. Both intracta- T-type voltage-gated calcium channels is heavily expressed ble pain and loss of sensation have significant adverse effects in the DRG cells and dorsal horn (DH) of the spinal cord on quality-of-life measures. Unfortunately, current treatment and plays a distinct role in supporting pathological pain in COMMENTARY options are unable to reverse these symptoms. animal models of PDN induced by both type 1 and type 2 Pain-sensing sensory neurons, or nociceptors, can be diabetes (4–6). Additional studies have documented the sensitized (become hyperexcitable) by various mecha- upregulation of pronociceptive ion channels (such as pu- nisms in response to the pathological conditions or pe- rinergic receptors [7]; voltage-gated sodium channels, partic- ripheral tissue injury associated with diabetes. -

4 Voltage-Gated Potassium Channels
SVNY290-Chung July 25, 2006 14:46 4 Voltage-Gated Potassium Channels Stephen J. Korn and Josef G. Trapani One change has been made and is noted. Part I. Overview Au: Please Potassium (K+) channels are largely responsible for shaping the electrical behavior check the relevance of of cell membranes. K+ channel currents set the resting membrane potential, control Part titles in action potential duration, control the rate of action potential firing, control the spread this chapter. 2 of excitation and Ca + influx, and provide active opposition to excitation. To support Should these these varied functions, there are a large number of K+ channel types, with a great be allowed? deal of phenotypic diversity, whose properties can be modified by many different Please accessory proteins and biochemical modulators. confirm. Also, As with other ion channels, there are two components to K channel opera- note that the + author has tion. First, channels provide a pathway through the cell membrane that selectively mentioned allows a particular ion species (in this case, K+) to flow with a high flux rate. Second, about the channels have a gating mechanism in the conduction pathway to control current flow copyright in response to an external stimulus. To accommodate their widespread involvement issues in a in cellular physiology, K channels respond to a large variety of stimuli, includ- para after the + Acknowledg- ing changes in membrane potential, an array of intracellular biochemical ligands, ments section, temperature, and mechanical stretch. Additional phenotypic variation results from which is a wide range of single-channel conductances, differences in stimulus threshold, and deleted here. -

Targeting of Voltage-Gated Potassium Channel Isoforms to Distinct Cell Surface Microdomains
Research Article 2155 Targeting of voltage-gated potassium channel isoforms to distinct cell surface microdomains Kristen M. S. O’Connell1 and Michael M. Tamkun1,2,* 1Department of Biomedical Sciences and 2Department of Biochemistry and Molecular Biology, Colorado State University, Ft Collins, CO 80523, USA *Author for correspondence (e-mail: [email protected]) Accepted 23 February 2005 Journal of Cell Science 118, 2155-2166 Published by The Company of Biologists 2005 doi:10.1242/jcs.02348 Summary Voltage-gated potassium (Kv) channels regulate action diffused throughout the cell surface. Additionally, PA- potential duration in nerve and muscle; therefore changes GFP-Kv2.1 moved into regions of the cell membrane not in the number and location of surface channels can adjacent to the original photoactivation ROI. Sucrose profoundly influence electrical excitability. To investigate density gradient analysis indicated that half of Kv2.1 is trafficking of Kv2.1, 1.4 and 1.3 within the plasma part of a large, macromolecular complex while Kv1.4 membrane, we combined the expression of fluorescent sediments as predicted for the tetrameric channel complex. protein-tagged Kv channels with live cell confocal imaging. Disruption of membrane cholesterol by cyclodextrin Kv2.1 exhibited a clustered distribution in HEK cells minimally altered Kv2.1 mobility (Mf=0.32±0.03), but similar to that seen in hippocampal neurons, whereas significantly increased surface cluster size by at least Kv1.4 and Kv1.3 were evenly distributed over the plasma fourfold. By comparison, the mobility of Kv1.4 decreased membrane. Using FRAP, surface Kv2.1 displayed limited following cholesterol depletion with no change in surface mobility; approximately 40% of the fluorescence recovered distribution. -

Prediction of Conotoxin Type Based on Long Short- Term Memory Network
Prediction of Conotoxin Type Based on Long Short- term Memory Network Feng Wang Changzhou University Huaide College Shan Chang Jiangsu University of Technology Dashun Wei ( [email protected] ) Huaide College of Changzhou University Research Keywords: Conotoxin, LSTM, prediction Posted Date: March 30th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-273779/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/12 Abstract Background: Conotoxin is a valuable peptide that targets ion channels and neuronal receptors. The toxin has been proven to be an effective drug for treating a series of diseases, but the process of identifying the type of toxin through traditional wet experiments is very complicated, low eciency and high cost, but the method of machine learning is used to identify the cono toxin. Training in the process can effectively change this status quo. Methods: A method to predict the type of spiral toxin using the sequence information of the toxin combined with the long-term short-term memory network (LSTM) method model. This method only needs to take the conotoxin peptide sequence as input, and uses the character embedding method in text processing to automatically map the sequence to the feature vector representation, and extract the features for training and prediction. Results: Experimental results show that the correct index of this method on the test set reaches 0.80, and the AUC (area under the ROC curve) value reaches 0.817. For the same test set, the AUC value of the KNN algorithm is 0.641, and the AUC value of the method proposed in this paper is 0.817. -

Cold Sensing by Nav1.8-Positive and Nav1.8-Negative Sensory Neurons
Cold sensing by NaV1.8-positive and NaV1.8-negative sensory neurons A. P. Luiza, D. I. MacDonalda, S. Santana-Varelaa, Q. Milleta, S. Sikandara, J. N. Wooda,1, and E. C. Emerya,1 aMolecular Nociception Group, Wolfson Institute for Biomedical Research, University College London, London WC1E 6BT, United Kingdom Edited by Peter McNaughton, King’s College London, London, United Kingdom, and accepted by Editorial Board Member David E. Clapham January 8, 2019 (received for review August 23, 2018) The ability to detect environmental cold serves as an important define the distribution and identity of cold-sensitive DRG neurons, survival tool. The sodium channels NaV1.8 and NaV1.9, as well as in live mice, using in vivo imaging. the TRP channel Trpm8, have been shown to contribute to cold sensation in mice. Surprisingly, transcriptional profiling shows that Results NaV1.8/NaV1.9 and Trpm8 are expressed in nonoverlapping neuro- Distribution of DRG Sensory Neurons Responsive to Noxious Cold, in nal populations. Here we have used in vivo GCaMP3 imaging to Vivo. To identify cold-sensitive neurons in vivo, we used a pre- identify cold-sensing populations of sensory neurons in live mice. viously developed in vivo imaging technique to study the responses We find that ∼80% of neurons responsive to cold down to 1 °C do of individual DRG neurons in situ (8). Mice coexpressing Pirt- not express NaV1.8, and that the genetic deletion of NaV1.8 does GCaMP3 (which enables pan-DRG GCaMP3 expression), not affect the relative number, distribution, or maximal response NaV1.8 Cre, and a Cre-dependent reporter (tdTomato) were of cold-sensitive neurons. -

Conotoxins As Tools to Understand the Physiological Function of Voltage-Gated Calcium (Cav) Channels
marine drugs Review Conotoxins as Tools to Understand the Physiological Function of Voltage-Gated Calcium (CaV) Channels David Ramírez 1,2, Wendy Gonzalez 1,3, Rafael A. Fissore 4 and Ingrid Carvacho 5,* 1 Centro de Bioinformática y Simulación Molecular, Universidad de Talca, 3460000 Talca, Chile; [email protected] (D.R.); [email protected] (W.G.) 2 Instituto de Ciencias Biomédicas, Universidad Autónoma de Chile, 3460000 Talca, Chile 3 Millennium Nucleus of Ion Channels-Associated Diseases (MiNICAD), Universidad de Talca, 3460000 Talca, Chile 4 Department of Veterinary and Animal Sciences, University of Massachusetts, Amherst, MA 01003, USA; rfi[email protected] 5 Department of Biology and Chemistry, Faculty of Basic Sciences, Universidad Católica del Maule, 3480112 Talca, Chile * Correspondence: [email protected]; Tel.: +56-71-220-3518 Received: 8 August 2017; Accepted: 4 October 2017; Published: 13 October 2017 Abstract: Voltage-gated calcium (CaV) channels are widely expressed and are essential for the completion of multiple physiological processes. Close regulation of their activity by specific inhibitors and agonists become fundamental to understand their role in cellular homeostasis as well as in human tissues and organs. CaV channels are divided into two groups depending on the membrane potential required to activate them: High-voltage activated (HVA, CaV1.1–1.4; CaV2.1–2.3) and Low-voltage activated (LVA, CaV3.1–3.3). HVA channels are highly expressed in brain (neurons), heart, and adrenal medulla (chromaffin cells), among others, and are also classified into subtypes which can be distinguished using pharmacological approaches. Cone snails are marine gastropods that capture their prey by injecting venom, “conopeptides”, which cause paralysis in a few seconds. -

Kinase and Phosphatase Activities Intimately Associated with a Reconstituted Calcium-Dependent Potassium Channel
The Journal of Neuroscience, June 1995, 15(6): 4572-4579 Kinase and Phosphatase Activities Intimately Associated with a Reconstituted Calcium-Dependent Potassium Channel Peter H. Reinhart and Irwin B. Levitan ‘Department of Neurobiology, Duke University Medical Center, Durham, North Carolina 27710 and *Biochemistry Department and Center for Complex Systems, Brandeis University, Waltham, Massachusetts 02254 Type-2 calcium-dependent potassium (K,,) channels from channels from the mammalian brain (Reinhart et al.. 1991; mammalian brain, reconstituted into planar phospholipid Chung et al., 1991). Specific functions of these channels in the bilayers, are modulated by ATP or ATP analogs via an en- nervous system include the setting of neuronal tiring frequencies dogenous protein kinase activity intimately associated with (Lancaster et al., 1991), participation in neuropeptide secretion the channel (Chung et al., 1991). We show here that the (White et al., 1991; Bielefeldt and Jackson, 1994a), and regu- endogenous protein kinase activity is protein kinase C lation of presynaptic calcium signals and neurotransmitter re- (PKC)-like because (1) modulation by ATP can be mimicked lease (Robitaille and Charlton, 1992). Using planar bilayer re- by exogenous PKC, and (2) the effects of ATP can be constitution techniques, we have identified two distinct types of blocked by PKCt,9.36j, a specific peptide inhibitor of PKC. large conductance K,.,, channel in membrane vesicles from rat Furthermore, adding the PKC inhibitor peptide after the ad- cortex (Reinhart et al., 1989). Channels with similar properties dition of ATP reverses the modulation produced by ATP, have also been identified using patch-clamp techniques (Wang suggesting that there is a phosphoprotein phosphatase ac- et al., 1992; Bielefeldt and Jackson, 1994a). -

Voltage-Gated Sodium Channel (Nav )
Voltage-gated sodium channel (NaV) protein dissection creates a set of functional pore-only proteins David Shayaa, Mohamed Kreirb, Rebecca A. Robbinsc, Stephanie Wonga, Justus Hammona, Andrea Brüggemannb, and Daniel L. Minor, Jr.a,c,d,e,f,1 aCardiovascular Research Institute, cDepartments of Biochemistry and Biophysics and dCellular and Molecular Pharmacology, eCalifornia Institute for Quantitative Biomedical Research, University of California, San Francisco, CA 94158-9001; fPhysical Biosciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720; and bNanion Technologies GmbH, Gabrielenstrasse 9, D-80636 Munich, Germany Edited by Richard W. Aldrich, University of Texas at Austin, Austin, TX, and approved June 14, 2011 (received for review April 30, 2011) Many voltage-gated ion channel (VGIC) superfamily members microscopy studies (19 Å) that have suggested some general contain six-transmembrane segments in which the first four form features of NaVs purified from eel electric organs (11) but that a voltage-sensing domain (VSD) and the last two form the pore lack the resolution for detailed mechanistic insight. Study of bac- domain (PD). Studies of potassium channels from the VGIC super- terial NaVs provides a simplified system for understanding basic family together with identification of voltage-sensor only proteins aspects of both NaV and CaV function and holds promise as a have suggested that the VSD and the PD can fold independently. template for guiding small molecule modulator development (6). Whether such transmembrane modularity is common to other Although there have been ongoing efforts to produce bacterial VGIC superfamily members has remained untested. Here we show, NaV samples that can be used for biochemical and structural using protein dissection, that the Silicibacter pomeroyi voltage- studies (12–14), these have yet to achieve the multi-milli-gram gated sodium channel (NaVSp1) PD forms a stand-alone, ion selec- amounts required for extensive structural studies. -

Disease Associated Mutations in KIR Proteins Linked to Aberrant Inward Rectifier Channel Trafficking
biomolecules Review Disease Associated Mutations in KIR Proteins Linked to Aberrant Inward Rectifier Channel Trafficking 1, 2, 2 1 Eva-Maria Zangerl-Plessl y, Muge Qile y, Meye Bloothooft , Anna Stary-Weinzinger and Marcel A. G. van der Heyden 2,* 1 Department of Pharmacology and Toxicology, University of Vienna, 1090 Vienna, Austria; [email protected] (E.-M.Z.-P.); [email protected] (A.S.-W.) 2 Department of Medical Physiology, Division of Heart & Lungs, University Medical Center Utrecht, 3584 CM Utrecht, The Netherlands; [email protected] (M.Q.); [email protected] (M.B.) * Correspondence: [email protected]; Tel.: +31-887558901 These authors contributed equally to this work. y Received: 28 August 2019; Accepted: 23 October 2019; Published: 25 October 2019 Abstract: The ubiquitously expressed family of inward rectifier potassium (KIR) channels, encoded by KCNJ genes, is primarily involved in cell excitability and potassium homeostasis. Channel mutations associate with a variety of severe human diseases and syndromes, affecting many organ systems including the central and peripheral neural system, heart, kidney, pancreas, and skeletal muscle. A number of mutations associate with altered ion channel expression at the plasma membrane, which might result from defective channel trafficking. Trafficking involves cellular processes that transport ion channels to and from their place of function. By alignment of all KIR channels, and depicting the trafficking associated mutations, three mutational hotspots were identified. One localized in the transmembrane-domain 1 and immediately adjacent sequences, one was found in the G-loop and Golgi-export domain, and the third one was detected at the immunoglobulin-like domain. -
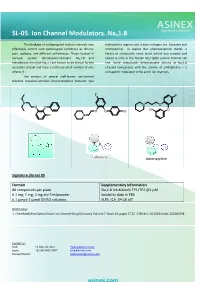
SL-05. Ion Channel Modulators. Nav1.8
SL-05. Ion Channel Modulators. Nav1.8 The blockage of voltage-gated sodium channels may hydrophobic regions and a basic nitrogen (i.e. lidocaine and effectively control such pathological conditions as chronic amitriptyline). To exploit this pharmacophore model, a pain, epilepsy, and different arrhythmias. Those located in library of structurally novel biaryl ethers was created and nervous system tetrodotoxin-resistant Nav1.8 and tested in vitro in the human Nav1.8/β3 sodium channel cell tetrodotoxin-sensitive Nav1.7 are known to be critical for the line. Some compounds demonstrated activity at Nav1.8 sensation of pain and have a relatively small number of side channel comparable with the activity of amitriptyline – a effects [1] nonspecific modulator of Na and K ion channels. The analysis of several well-known ion-channel blockers revealed common pharmacophoric features: two F F N N O O O Cl O O O Cl F N Lidocaine Amitriptyline Signature Library 05 Formats Supplementary Information 80 compounds per plate Nav1.8 Inhibition% TP1/TP2 @1 μM 0.1 mg; 1 mg; 2 mg dry film/powder Solubility data in PBS 0.1 µmol; 1 µmol DMSO solutions SL#5_ICh_04-16.sdf References: 1. ChemMedChem Special Issue: Ion Channel Drug Discovery Volume 7, Issue 10, pages 1712–1740 doi: 10.1002/cmdc.201200298. Contact us: USA: +1 336 721 1617 [email protected] Japan: +81-80-3401-9097 [email protected] Europe/Global: [email protected] SL-51. Nav1.8 Channel Modulators Voltage-gated sodium channels are involved in has been used to carry out an exhaustive search on a 20K+ signal transduction in electrically excitable tissues such as set of natural product-like compounds.