Development of Simple and Efficient Synthetic Strategies For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Technical Information
Technical Information Antibiotic Assay Medium No.1 (Seed Agar) Product Code : DM1003 Application: - Antibiotic Assay Medium No.1 (Seed Agar) is used in the microbiological assay of beta-lactam and other antibiotics. Composition** Ingredients Gms / Litre Peptic digest of animal tissue (Peptone) 6.000 or detecting faecal coliforms drinking in water waste water, seawater and foods samples by MPN Method. Casein enzymic hydrolysate 4.000 Yeast extract 3.000 Beef extract 1.500 Dextrose 1.000 Agar 15.000 Final pH (at 25°C) 6.6±0.2 **Formula adjusted, standardized to suit performance parameters Principle & Interpretation The potency of an antibiotic can be determined by chemical, physical and biological methods. An assay is performed to determine the ability of an antibiotic to kill or inhibit the growth of living microorganisms. Biological tests offer the most convenient m eans of performing an assay (1), since a reduction in the antimicrobial activity of a specific antibiotic reveals changes that is not usually displayed by chemical methods (2). Antibacterial susceptibility testing may be performed by either dilution (turbidimetric) or diffusion methods. The choice o f methodology is based on many factors, including ease of performance, flexibility and use of automated or semi-automated devices for both identification and susceptibility testing (3). Grove and Randall have elucidated antibiotic assays and media in their comprehensive treatise on antibiotic assays (4). Antibiotic Assay Medium No.1 is used in the microbiological assay of ß-lactam and other antibiotics. These media are prepared according to the specifications detailed in various pharmacopoeias (2-6) and by the FDA (7). -

Contractor Orientation Manual
Contractor Orientation Manual KALAMAZOO CONTRACTOR ADMINISTRATION TABLE OF CONTENTS INTRODUCTION......................................................................................................................................4 DEFINITIONS...........................................................................................................................................4 1. Supplier: ........................................................................................................................................4 2. Contractor: ………………………………………………………………………………………4 3. Pfizer Contact: ...…………………………………………………………………………….4 4. Kalamazoo Contractor Administration: ……………………………………………………..4 GENERAL INFORMATION....................................................................................................................5 1. Company Access ...........................................................................................................................5 2. Vehicle Admittance & Parking .....................................................................................................5 3. Tobacco Products ..........................................................................................................................6 4. Cafeterias and Snack Bars..............................................................................................................6 5. The Federal Food, Drug and Cosmetic Act................................................................................... 6 6. Good Manufacturing Practices -

Custom Manufacturing in Thailand, Evonik in China
10/2011 October Markets and Companies Markets and Companies Interview with Styrolution’s CEO THE NEWSPAPER FOR THE Expert Peter Pollak discusses the Roberto Gualdoni CHEMICAL AND fragmented world of fine chemicals Page 5 LIFE SCIENCE MARKE TS Pages 8 and 14 N EWSFLOW Market and Companies: Haltermann sold to H.I.G Europe, Dr. Uwe Nickel named new CEO. An Industry Full of Opportunity DuPont seeking buyers for two se- parate businesses: a polyester-film CPhI Highlights the Latest Trends in Pharma joint venture and one that makes powder-based paint. Bayer and Yunona Holdings have entered a memorandum of under- Markus Blocher Dr. Jörn Winterfeld standing to manufacture drugs in CEO, Dottikon Russia. Director Business Line Pharma/Agro at Wacker Biosolutions, Wacker AstraZeneca has announced it will be laying off about 400 employees in the U.S., primarily at its head- API Fine Chemicals quarters in Wilmington, Del. … Chemical exclusive synthesis partners Consolidation in the fine chemicals industry Lanxess has announced three in- need to possess a versatile technology will continue, since it is a very fragmented vestments totaling upwards of €30 “ portfolio like a Swiss army knife and be as industry. Many small players may vanish million in Brazil. ” “ ” precise and reliable as a Swiss watch. or merge to attain the so-called critical mass. Syngenta has posted a 21 % rise in This trend includes producers in China. its Q3 results over last year. More on Page 2 ▶ Jean Bléhaut Director of Marketing & Business Development, Novasep Dr. Martin Wienkenhöver Under Construction: CEO, CABB Both Solvay and Evonik are building hydrogen peroxide plants; Solvay Custom Manufacturing in Thailand, Evonik in China. -

Evolution of a Regulatory Framework for Pharmaceuticals Derived from Genetically Modified Plants
Review Evolution of a regulatory framework for pharmaceuticals derived from genetically modified plants Armin Spo¨k1*, Richard M. Twyman2, Rainer Fischer3, Julian K.C. Ma4 and Penelope A.C. Sparrow5* 1 Inter-University Research Centre for Technology, Work and Culture (IFZ), Schlo¨ gelgasse 2, A-8010 Graz, Austria 2 Department of Biology, University of York, Heslington, York, YO10 5DD, UK 3 Fraunhofer Institute for Molecular Biology and Applied Ecology (IME), Aachen, Germany 4 St George’s Hospital Medical School, Cranmer Terrace, London, SW17 0RE, UK 5 John Innes Centre, Norwich Research Park, Norwich, NR4 7UH, UK The use of genetically modified (GM) plants to synthes- phase II clinical trials, respectively [6]. In 2006, the United ize proteins that are subsequently processed, regulated States Department of Agriculture (USDA) licensed a poul- and sold as pharmaceuticals challenges two very differ- try vaccine produced in cultured tobacco cells [7]. Since ent established regulatory frameworks, one concerning then, several products derived from crop plants have also GM plants and the other covering the development of reached late development stages, including human insulin biotechnology-derived drugs. Within these regulatory and carp growth hormone produced in safflower. These are systems, specific regulations and guidelines for plant- expected to reach the market between 2008 and 2010 (see made pharmaceuticals (PMPs) – also referred to as plant- Table 1). derived pharmaceuticals (PDPs) – are still evolving. The PMPs present two major challenges for the regulatory products nearing commercial viability will ultimately bodies. Regulators of agricultural biotechnology are con- help to road test and fine-tune these regulations, and fronted with a novel type of crop use, and drug regulators might help to reduce regulatory uncertainties. -

New Deals, New Investments in Chemical Manufacture
Chemical Manufacture New deals, new investments in chemical manufacture We review some of the most recent contracts, investments and merger and acquisition activities in the highly competitive field of fine chemical manufacture. he manufacture of pharmaceutical fine chemical business is a major player in the global fine chemical sector, intermediates and ingredients remains probably the with a leading position in the supply of contract manufacturing Tmost competitive sector of the chemical industry and services to the agrochemicals, pharmaceutical and speciality there continue to be major changes in the structure of the chemical industries. The acquisition included all Avecia Fine sector and in the geographical spread of major manufacturing Chemicals’ assets and operations on the 65 hectare site at faciltiies, with investments in China and India playing a leading Grangemouth, UK. All 310 Avecia Fine Chemicals’ employees part in companies’ moves to acquire stronger positions. This were included in the transaction. In 2004, the business article looks at just a few of the recent developments. recorded sales of £38 million. Avecia Fine Chemicals Limited now operates as KemFine UK Acquisition of ozone chemistry technology Ltd. Tom Shields is the managing director, and is based at the In February, Dishman Pharmaceuticals & Chemicals Ltd Grangemouth site. KemFine, previously Kemira Fine Chemicals acquired IO3S Ltd of Bern, Switzerland, through its 100% Oy, was created via an MBO of the business from Kemira in wholly owned Switzerland based subsidiary company M/s. 2004. 3i, a world leader in the private equity and venture Dishman Switzerland Ltd. The Assets base of IO3S is currently capital sector, supported the management team in the about $2 million but Dishman says the acquisition was transaction and in the equity funding. -

Chemacx Vendor List
ChemACX Vendor List -A B C D E F G H I J K L M N O P Q R S T U V W X Y Z- -#- APAC PHARMACEUTICAL ACTIVATE SCIENTIFIC GMBH AQCHEM LLC ADESIS INC. 1K SCIENTIFIC ASW MEDCHEM, INC. ADVAMACS 3A PHARMATECH AURUM PHARMATECH LLC ADVANCE RESEARCH CHEMICALS, INC. 3B SCIENTIFIC CORP. AARON CHEMISTRY ADVANCED AROMATICS, L.P. 3RCHEM ABAMACHEM LTD ADVANCED ASYMMETRICS, INC. 8S LABORATORIES LLC ABBEY COLOR ADVANCED CHEMBLOCKS INC. Updated Aug. 2018 -A- ABBOTT LABORATORIES CHEMICAL DIVISION ADVANCED CHEMTECH A&A LIFE SCIENCE INC. DBA ABBY PHARMATECH LLC ADVANCED MOLECULAR AVACHEM SCIENTIFICS TECHNOLOGIES PTY LTD ABERJONA LABORATORIES A&C PHARMTECH, INC ADVANCED SCIENTIFIC ABOVCHEM LLC INTERNATIONAL, LLC A&J PHARMATECH ABSOLUTE CHIRAL ADVANCED SYNTHESIS A.G. SCIENTIFIC Updated July 2018 TECHNOLOGIES, S.A. Updated Jan. 2018 ACADECHEM COMPANY LIMITED ADVANCED TECHNOLOGY & A1 BIOCHEM LABS INDUSTRIAL CO., LTD. Updated Sep. 2018 ACADEMIQUE PHYTOCANADA AIMS FINE CHEMICALS INC. AA BLOCKS LLC ACCELA CHEMBIO CO., LTD. New Sep. 2018 Updated June 2018 AIR PRODUCTS & CHEMICALS, INC. AB CHEM TECHNOLOGIES LLC ACCELEDEV CHEMICAL LLC AJINOMOTO - AMINO ACID DEPARTMENT ABCR GMBH & CO. KG ACCUSTANDARD, INC. ALBANY MOLECULAR RESEARCH, INC. ABSCO LTD ACE SYNTHESIS LLC ALBEMARLE CORPORATION ABX ADVANCED BIOCHEMICAL ACENTEX SCIENTIFIC, INC. COMPOUNDS ALBRIGHT & WILSON AMERICAS ACESYS PHARMATECH AECHEM ALCHEM INTERNATIONAL LIMITED ACETO CORPORATION AK SCIENTIFIC, INC. ALCHEM PHARMTECH, INC. Updated Sep. 2018 ACHEMICA ALDLAB CHEMICALS, LLC AKL RESEARCH LLP ACINOPEPTIDE CO. LTD ALDRICH AOBCHEM USA ACME BIOSCIENCE, INC. ALFA AESAR AOKBIO CO., LTD ACROS ORGANICS Updated Apr. 2018 ALFA CHEMISTRY ACROS ORGANICS – USA New June 2018 AOKCHEM CO., LTD. -

A Fine Art How Much of the Fin E Chemicals Market Can Pharma Lay Claim to Today- and in the Future? Jan Ramakers, Jan Ramakers Fine Chemical Consulting Group Writes
E CHEMICALS • A fine art How much of the fin e chemicals market can pharma lay claim to today- and in the future? Jan Ramakers, Jan Ramakers Fine Chemical Consulting Group writes ine chemicals, speciality chemicals, and performance chemicals are some of the various names used to describe products in the Fine chemical market in 2014 Fhigh-value end of the chemical market. In order to avoid any - total value $127.7bn - confusion, it is good to keep in mind that fine chemicals ore defined as (high value) chemical products that ore sold for what they ore, ie for Other Pharma their precise chemical structure. Speciality chemicals, often also referred Food a to as performance chemicals, are (high value) chemical products that 4% ore sold for what they do. So an intermediate used in the manufacture Pigments& of a specific API is a fine chemical as it needs to have a very specific 3% chemical structure; a different chemical structure cannot be used. Fine chemicals are used across a wide range of market segments, and it is easy enough to make a list that features 40-50 different application intermediates areas. 10% The products end up in speciality chemicals markets, for instance as Fragrances active ingredients in biocide formulations, as additives for plastics and 7% coatings, or as active ingredients in cosmetics and toiletries. Some are Figure 1. used as intermediates in the manufacture of liquid crystal displays, and so on. Many of those end markets are quite large, but most of them are fairly small as an outlet for fine chemicals. -

(12) Patent Application Publication (10) Pub. No.: US 2007/0122887 A1 Klopprogge Et Al
US 2007.0122887A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2007/0122887 A1 Klopprogge et al. (43) Pub. Date: May 31, 2007 (54) GENE VARIANTS CODING FOR PROTEINS (30) Foreign Application Priority Data FROM THE METABOLIC PATHWAY OF FNE CHEMICALS Dec. 18, 2003 (DE)..................................... 103 59 661.5 (75) Inventors: Corinna Klopprogge, Mannheim (DE); Publication Classification Oskar Zelder, Speyer (DE); Burkhard Kroger, Limburgerhof (DE); Hartwig (51) Int. Cl. Schroder, Nublock (DE); Stefan CI2P I3/04 (2006.01) Haefner, Ludwigshafen (DE); Uwe CI2P I3/08 (2006.01) Ruffer, Meckenheim (DE); Claudia C7H 2L/04 (2006.01) Isabella Graef, Lampertheim (DE) CI2N 9/10 (2006.01) CI2N 15/74 (2006.01) Correspondence Address: CI2N I/2 (2006.01) LAHIVE & COCKFIELD, LLP ONE POST OFFICE SQUARE (52) U.S. Cl. ......................... 435/106; 435/115; 435/193; BOSTON, MA 02109-2127 (US) 435/252.3; 435/471; 536/23.2 (73) Assignee: BASF Aktiengesellschaft, Ludwigshafen (DE) (57) ABSTRACT (21) Appl. No.: 10/583,404 (22) PCT Filed: Dec. 16, 2004 The present invention relates to mutated nucleic acids and proteins of the metabolic pathway of fine chemicals, to (86). PCT No.: PCT/EPO4/14338 processes for preparing genetically modified producer organisms, to processes for preparing fine chemicals by S 371(c)(1), culturing said genetically modified organisms and to said (2), (4) Date: Jun. 16, 2006 genetically modified organisms themselves. US 2007/O 122887 A1 May 31, 2007 GENEVARLANTS CODNG FOR PROTEINS FROM and optimized C yield, there is a constant need for further THE METABOLIC PATHWAY OF FINE improving the productivity of said organisms. -

Fine Chemical Intermediates
Fine Chemical Intermediates On Request (OR) / Main Structure Identification Characteristics Regular Applications Production (RP) Phosgene derivatives Methyl chloroformate Assay: min. 99% by area Organic reagent for RP O MCF, Methyl chlorocarbo- introduction of the nate, Chloroformic acid Appearance: colourless to methoxycarbonyl methyl ester pale yellow liquid group, synthesis of Packaging: 160 kg drum, carbonates, Cl O CAS Reg. No. 79-22-1 ISO tank carbamates, anhydrides etc. Ethyl chloroformate Assay: min. 98.5% by area Organic reagent for RP O ECF, Ethyl introduction of the chlorocarbonate, Appearance: colourless to ethoxycarbonyl group, Chloroformic acid ethyl pale yellow liquid synthesis of ester Packaging: 160 kg drum, carbonates, Cl O ISO tank carbamates, CAS Reg. No. 541-41-3 anhydrides etc. n-Propyl chloroformate Assay: min. 98% by area Organic reagent for OR O nPCF, n-Propyl chlorocar- introduction of the n- bonate, Chloroformic acid Appearance: colourless to propoxycarbonyl propyl ester pale yellow liquid group, synthesis of Cl O Packaging: ISO tank carbonates, CAS Reg. No. 109-61-5 carbamates, anhydrides etc. Isopropyl chloroformate Assay: min. 98.5% by area Organic reagent for OR O iPCF, 2- introduction of the Propylchloroformate, Appearance: clear colour- isopropoxycarbonyl Chloroformic acid less to pale yellow liquid group, synthesis of isopropyl ester Packaging: ISO tank carbonates, Cl O carbamates, CAS Reg. No. 108-23-6 anhydrides etc. n-Pentylchloroformate Assay: min. 98.5% by area Organic reagent for OR n-Pentylchlorocarbonate, introduction of the n- O Chloroformic acid pentyl Appearance: colourless pentoxycarbonyl ester liquid group, synthesis of Packaging: 190 kg drum carbonates, Cl O CAS Reg. No. 638-41-5 carbamates, anhydrides etc. -

Cbn Smis Intervention of March 2, 2017 - Sales
CBN SMIS INTERVENTION OF MARCH 2, 2017 - SALES S/N CUSTOMERS NAME AMOUNT (US$) RATE (N/US$) ITEM OF IMPORT FORM 'M' NUMBER LC NUMBER DATE OPENED MATURITY DATE BANK 45- DAY TENOR 1 SMSAM SYSTEM LTD 20,500.00 380.00 SOFTWARE MF20160100500 22157280377 UNITY 2 PROCTER AND GAMBLE NIGERIA LIMITED 52,623.11 375.00 KRAFT BLEACHED WOODPULP 25.0CM MF20160086965X 152.0CM 516 (RAW MATERIAL5067600865 FOR PRODUCING7/28/16 PAMPERS) 1/17/17 CITI 3 PROCTER AND GAMBLE NIGERIA LIMITED 92,391.70 375.00 KRAFT BLEACHED WOODPULP CMC530MF20160086945 (RAW MATERIAL FOR PRODUCING5063600865 PAMPERS)7/28/16 10/28/16 CITI 4 PROCTER AND GAMBLE NIGERIA LIMITED 89,851.22 375.00 BG PA BFLY S2 10 SMP SSA; BG PA SF BFLYMF20160095066 S2 80 JP SSA; BG PA SF 5064600890BFLY S4 64 JP8/16/16 SSA; BG PA SF BFLY S3 911/4/16 SMP SSA; BG PA SF BFLY S3CITI 72 JP SSA (RAW MATERIAL FOR PRODUCING PAMPERS) 5 PROCTER AND GAMBLE NIGERIA LIMITED 214,060.00 375.00 AQUALIC CA L521 (RAW MATERIAL FORMF20160091918 PRODUCING PAMPERS) 5069600877 8/8/16 11/28/16 CITI 6 PROCTER AND GAMBLE NIGERIA LIMITED 183,480.00 375.00 AQUALIC CA L521 (RAW MATERIAL FORMF20160094963 PRODUCING PAMPERS) 5063600890 8/16/16 11/28/16 CITI 7 PROCTER AND GAMBLE NIGERIA LIMITED 53,243.14 375.00 KRAFT BLEACHED WOODPULP 50.8CM MF20160095724X 120.0CM 516 (RAW MATERIAL5062600891 FOR PRODUCING8/17/16 PAMPERS) 10/28/16 CITI 8 PROCTER AND GAMBLE NIGERIA LIMITED 61,537.41 375.00 POLYESTER NONWOVEN ACQUISITIONMF20160093058 LAYER (43G/M2) AQL 2+ WHITE5061600876 80 MM (RAW8/8/16 MATERIALS FOR PRODUCING1/2/17 PAMPERS) CITI 9 PROCTER AND GAMBLE NIGERIA LIMITED 42,302.05 375.00 980 - U- TAPES 13MM WIDTH 13MM ADHMF20160086914 U GLUE 8 BOX/FUM. -
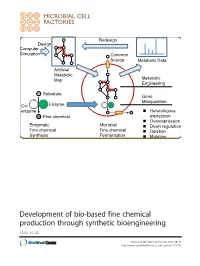
Development of Bio-Based Fine Chemical Production Through Synthetic Bioengineering Hara Et Al
Redesign Design Computer Simulation Common Source Metabolic Data Artificial Metabolic Map Metabolic Engineering Substrate Gene Manipulation Co- Enzyme enzyme Heterologous Fine chemical expression Overexpression Enzymatic Microbial Down regulation Fine chemical Fine chemical Deletion Synthesis Fermentation Mutation Development of bio-based fine chemical production through synthetic bioengineering Hara et al. Hara et al. Microbial Cell Factories 2014, 13:173 http://www.microbialcellfactories.com/content/13/1/173 Hara et al. Microbial Cell Factories (2014) 13:173 DOI 10.1186/s12934-014-0173-5 REVIEW Open Access Development of bio-based fine chemical production through synthetic bioengineering Kiyotaka Y Hara1, Michihiro Araki1, Naoko Okai1, Satoshi Wakai1, Tomohisa Hasunuma1 and Akihiko Kondo2* Abstract Fine chemicals that are physiologically active, such as pharmaceuticals, cosmetics, nutritional supplements, flavoring agents as well as additives for foods, feed, and fertilizer are produced by enzymatically or through microbial fermentation. The identification of enzymes that catalyze the target reaction makes possible the enzymatic synthesis of the desired fine chemical. The genes encoding these enzymes are then introduced into suitable microbial hosts that are cultured with inexpensive, naturally abundant carbon sources, and other nutrients. Metabolic engineering create efficient microbial cell factories for producing chemicals at higher yields. Molecular genetic techniques are then used to optimize metabolic pathways of genetically and metabolically well-characterized hosts. Synthetic bioengineering represents a novel approach to employ a combination of computer simulation and metabolic analysis to design artificial metabolic pathways suitable for mass production of target chemicals in host strains. In the present review, we summarize recent studies on bio-based fine chemical production and assess the potential of synthetic bioengineering for further improving their productivity. -

Chemical Industry of the Future: New Biocatalysts
NEW BIOCATALYSTS: ESSENTIAL TOOLS FOR A SUSTAINABLE 21st CENTURY CHEMICAL INDUSTRY Sponsored by Altus Genencor International, Inc. Cargill, Inc. Chevron Council for Chemical Research DSM E.I. DuPont de Nemours & Co. Maxygen U.S. Department of Commerce, NIST/ATP U.S. Department of Defense, Office of Naval Research U.S. Department of Energy, Energy Efficiency and Renewable Energy, Office of Industrial Technologies Utah State University Biotechnology Center Facilitation by Energetics, Inc. Sponsors Council for Chemical Research ® Table of Contents 1 Section I. Executive Summary 3 Section II. Introduction 7 Section III. The Visioning Process and Drivers 9 Section IV. The Present State of Biocatalysts 13 Section V. The Workshop 19 Section VI. Implementation 27 Section VII. Success Factors in Implementing the R & D in the Roadmap 33 Appendix A 34 Appendix B 36 List of Participants 37 Contact Information 38 Acknowledgments 2 We wish to thank the sponsors listed on the front cover for their support, without which this workshop could not have taken place nor could this report have been written. Special thanks to Genencor for use of their facilities and to Jack Huttner, Richard LaDuca, Jim Sjoerdsma, and Nancy Shaw for their hospitality. The authors thank the planning group and participants in the workshop for their many suggestions and careful review of the several drafts. Special credit is due Brian Davison and David Boron for their contribu- tions and graphics. Technical assistance by Mary Donahue, graphic designer, and Georgia Herod, editor, was superb. Pamela Garcia served as the workshop secre- tariat and did an excellent job in a complex organizational task.