Copyright Undertaking
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Index Page 12-13(TD)-THB &
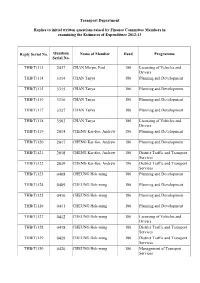
Transport Department Replies to initial written questions raised by Finance Committee Members in examining the Estimates of Expenditure 2012-13 Reply Serial No. Question Name of Member Head Programme Serial No. THB(T)113 2437 CHAN Mo-po, Paul 186 Licensing of Vehicles and Drivers THB(T)114 3314 CHAN Tanya 186 Planning and Development THB(T)115 3315 CHAN Tanya 186 Planning and Development THB(T)116 3316 CHAN Tanya 186 Planning and Development THB(T)117 3317 CHAN Tanya 186 Planning and Development THB(T)118 3503 CHAN Tanya 186 Licensing of Vehicles and Drivers THB(T)119 2015 CHENG Kar-foo, Andrew 186 Planning and Development THB(T)120 2017 CHENG Kar-foo, Andrew 186 Planning and Development THB(T)121 2018 CHENG Kar-foo, Andrew 186 District Traffic and Transport Services THB(T)122 2019 CHENG Kar-foo, Andrew 186 District Traffic and Transport Services THB(T)123 0408 CHEUNG Hok-ming 186 Planning and Development THB(T)124 0409 CHEUNG Hok-ming 186 Planning and Development THB(T)125 0410 CHEUNG Hok-ming 186 Planning and Development THB(T)126 0411 CHEUNG Hok-ming 186 Planning and Development THB(T)127 0412 CHEUNG Hok-ming 186 Licensing of Vehicles and Drivers THB(T)128 0418 CHEUNG Hok-ming 186 District Traffic and Transport Services THB(T)129 0419 CHEUNG Hok-ming 186 District Traffic and Transport Services THB(T)130 0420 CHEUNG Hok-ming 186 Management of Transport Services Reply Serial No. Question Name of Member Head Programme Serial No. THB(T)131 0541 CHEUNG Yu-yan, Tommy 186 Licensing of Vehicles and Drivers THB(T)132 0542 CHEUNG Yu-yan, Tommy -

Hong Kong's Bad Biodiversity
JULY 2001 NUMBER 23 Porcupine! Newsletter of the Department of Ecology & Biodiversity, The University of Hong Kong Hong Kong's Bad Biodiversity Aliens - also known as exotics or introduced species - are species which did not occur here naturally but have been brought to Hong Kong by humans, either deliberately, as crops, ornamentals, domestic animals or pets, or accidentally, as stowaways in ships and cargo planes, as seeds or eggs in soil, or attached to shoes or clothing. Most species which arrive in this way do not "escape" to form wild populations, and most of the species which have escaped are confined to places that are continually disturbed by humans. The weeds of cultivated areas, roadsides and urban wasteland are mostly exotics, as are the fish in concrete channels, and such conspicuous urban invertebrates as the "American" Cockroach and the Giant African Snail. Most of these urban exotics are doing no obvious harm and some contribute positively to the quality of city life. However, they also form the pool of species from which are drawn the small number of ecologically-damaging ones: the invasive species. Invasive species are aliens that invade natural or semi-natural communities. Out of the 150 or so well-established alien plant species in Hong Kong, less than a dozen have become invasive. The climbing American composite Mikania micrantha – Mile-a-Minute – is the most obvious example, but even this is confined to relatively disturbed sites. The "success" rate seems to be higher among birds and mammals, but the species which have done well in natural habitats are all from tropical East Asia and are apparently occupying niches vacated by the loss of the same or closely-related species when Hong Kong was deforested centuries ago. -

Recommended District Council Constituency Areas
District : Tsuen Wan Recommended District Council Constituency Areas +/- % of Population Estimated Quota Code Recommended Name Boundary Description Major Estates/Areas Population (17,282) K01 Tak Wah 20,527 +18.78 N Tai Ho Road North, Sai Lau Kok Road 1. CHUNG ON BUILDING 2. CITY LANDMARK I NE Sai Lau Kok Road 3. FOU WAH CENTRE E Sai Lau Kok Road, Shing Mun Road 4. HO FAI GARDEN 5. TAK YAN BUILDING (PART) : Kwan Mun Hau Street, Sha Tsui Road Stage 2 Chuen Lung Street, Ho Pui Street Stage 4 Chung On Street, Yeung Uk Road Stage 6 Stage 7 SE Ma Tau Pa Road, Texaco Road Stage 8 S Texaco Road, Tsuen Wan Road 6. THE BLUE YARD 7. THE DYNASTY SW Tsuen Wan Road, Tai Ho Road 8. TSUEN CHEONG CENTRE W Tai Ho Road 9. TSUEN WAN TOWN SQUARE NW Tai Ho Road, Tai Ho Road North 10. VISION CITY 11. WAH SHING BUILDING K1 District : Tsuen Wan Recommended District Council Constituency Areas +/- % of Population Estimated Quota Code Recommended Name Boundary Description Major Estates/Areas Population (17,282) K02 Yeung Uk Road 19,327 +11.83 N Sha Tsui Road, Kwan Mun Hau Street 1. BO SHEK MANSION 2. CHELSEA COURT Castle Peak Road - Tsuen Wan 3. EAST ASIA GARDENS NE Castle Peak Road - Tsuen Wan 4. HARMONY GARDEN 5. INDI HOME Texaco Road Flyover, Texaco Interchange 6. NEW HAVEN Texaco Road 7. TSUEN WAN GARDEN E Texaco Road, Texaco Road Flyover 8. WEALTHY GARDEN SE Texaco Road S Texaco Road, Tsuen Tsing Interchange Tsuen Wan Road SW Tsuen Tsing Interchange, Tsuen Wan Road W Texaco Road, Ma Tau Pa Road Yeung Uk Road, Chung On Street Ho Pui Street NW Chuen Lung Street K2 District : Tsuen Wan Recommended District Council Constituency Areas +/- % of Population Estimated Quota Code Recommended Name Boundary Description Major Estates/Areas Population (17,282) K03 Hoi Bun 18,826 +8.93 N Tai Ho Road, Tsuen Wan Road 1. -

Legislative Council
立法會 Legislative Council LC Paper No. CB(1)205/12-13(02) Ref: CB1/PL/TP Panel on Transport Special meeting on 27 November 2012 Background brief on safety of franchised bus operation and safety of long downhill roads Purpose This paper gives an account of the major views and concerns expressed by Legislative Council ("LegCo") Members and the Panel on Transport ("the Panel") in past discussion on the safety of franchised bus operation and safety of long downhill roads. Background 2. The Transport Department ("TD") monitors the operation of franchised bus services in accordance with the Public Bus Services Ordinance (Cap. 230) and the Road Traffic Ordinance ("RTO") (Cap. 374) and their Regulations. The franchised bus operators are required to carry out maintenance and repair as the Commissioner for Transport may specify, and TD's examiners are empowered to inspect the buses and maintenance facilities at any reasonable time. While buses should observe the general speed limits designated on roads, the maximum speed of a bus is restricted under RTO to 70 km/h on roads with a posted speed limit over 70km/h. 3. The Panel has all along been actively following the items on safety of long downhill roads and safety of franchised bus operation. The major incidents involving the operation of franchised buses and happening on long downhill roads are set out below. Traffic accident on Tuen Mun Road 4. Following a serious traffic accident happened on Tuen Mun Road on 10 July 2003, in which a bus carrying 40 passengers broke through a section of vehicular parapet and plunged into the hillside about 31 metres beneath, resulting in 21 fatalities and 20 injuries, the Chief Executive - 2 - appointed an independent Expert Panel to examine and make recommendations on safety measures to prevent similar catastrophes. -

The Proposal
THE PROPOSAL INTRODUCTION The Mountain Biking Explosion The exponential growth of recreational and sporting off road mountain bike riding is worldwide. Both cross country and Downhill Mountain Biking are established as Olympic events; the demand will con- tinue to increase. Locally The Hong Kong Magazine for August carried an article on mountain biking in Hong Kong, which only further fuels demand. Companies recruiting expatriates cite mountain biking and refer candidates to the HKMBA website as evidence that Hong Kong is a “cool” city to live. And in the latest Action Asia magazine there are three separate articles about mountain biking. Mountain Biking is a growing sport across the world, the bicycle industry in 2008 in the USA is a $6 billion industry, includ- ing the retail value of bicycles, related parts, and accessories according to research funded by the USA National Sporting Goods Association. Ironically the majority of bicycles, components and accessories are manufactured in China, and in Taiwan and Japan. Hong Kong’s bike shops are amongst the best in the world, when a new bike or component is manufac- tured it is usual to find it in a Hong Kong bike shop ahead of anywhere else. This is because most bikes and components are manufactured within the region and the Hong Kong bicycle enthusiast is one of the most discerning. Bicycle shops in other countries tend to carry minimum stock and will custom order Unfortunately for mountain biking cycling in the country parks falls under the authority of the AFCD. depending on requests by individual customers; recently online retailers overseas have filled this void by Whilst AFCD is no doubt qualified to fulfil their other responsibilities in the area of recreational use of offering huge catalogues and overnight delivery. -

(Cps), Transportation to the Finish Point
YOUR CARBON MANAGEMENT PARTNER Through our carbon footprint evaluation and advisory services, SGS is committed to assisting organizations in environmental compliance and achieving the goal on carbon reduction. SGS is proud to be the official Green Management Partner of Oxfam Trailwalker since 2009. SGS IS THE WORLD'S LEADING INSPECTION, VERIFICATION, TESTING AND CERTIFICATION COMPANY SGS Certification and Business Enhancement Phone: +852 2765 3620 | Email: [email protected] SGS Academy https://learning.sgs.com/hk www.sgsgroup.com.hk SGS-Oxfam-Sponsorship-2019.indd 1 20-May-19 11:35:27 AM Trailwalker Route The route starts at Pak Tam Chung P.H.A.B Site in Sai Kung, and covers sections 1 to 9 of the MacLehose Trail, which is about 88 km (M001 – M174). After passing distance post M174 and leaving CP9, the route leaves the MacLehose Trail and travels 5 km along a country trail to the north coast of Tai Lam Chung Reservoir. The trail then turns right along a paved road towards Wong Nai Tun/Tai Tong and heads towards the Finish Point at Tai Tong Barbecue Area in Yuen Long, the marker posts of OTW175 – OTW195 will be set up by the Organiser for identification. Yuen Long Tai Tong CP2 CP7 Finish CP8 CP3 CP1 CP9 CP6 CP4 Start CP5 Sai Kung CP = Checkpoint Map of Hong Kong MacLehose Trail Oxfam Trailwalker Route Section 2 M031-M033 From M031 pass through Sai Wan beach to M033 Section 8 M153-M156 From M153 along Tai Mo Shan Road to Route Twisk Section 10 M175-M200 From OTW175 to OTW195 (Turn left towards Kat Hing Bridge after registering at CP9, then walk along a country trail next to Tai Lam Chung Reservoir.) Participants should stay on the marked course. -

Trailwalker Route
TRAILWALKER ROUTE Route The 100 km route starts in Pak Tam Chung at Sai Kung, and covers sections 1 to 9 of the MacLehose Trail, which is about 88 km (M001 – M174). After passing distance post M174 and leaving CP9, the route leaves the MacLehose Trail and travels five kilometers along a country trail to the north coast of Tai Lam Chung Reservoir. The trail then turns right along a paved road towards Wong Nai Tun / Tai Tong and heads towards the Finish Point at the Po Leung Kuk Jockey Club Tai Tong Holiday Camp in Yuen Long (OTW175 – OTW198). Finish: Yuen Long (Tai Tong) Start: Sai Kung (Pak Tam Chung) CP = Checkpoint Important Note: The Oxfam Trailwalker trail is not identical to the MacLehose Trail. MacLehose Trail Oxfam Trailwalker route Section 2 M031-M033 From M031 pass through Sai Wan beach to M033 From M153 along Tai Mo Shan Road (takes the old MacLehose Trail) to Route Section 8 M153-M156 Twisk From OTW175 to OTW198 and the countdown signage inside the Finish Section 10 M175-M200 Point campsite (turn left towards Kat Hing Bridge after registering at CP9, then walk along a country trail next to Tai Lam Chung Reservoir) Participants should stay on the marked course. Shortcuts are strictly prohibited. The Organiser will carry out random checks. The Organiser will not record the results and rankings of participants who are found to have taken shortcuts. 27 28 Oxfarm_150mmx220mm.indd 1 18/4/2018 11:52 AM CHECKPOINT (CP) DISTANCES & ELEVATION CP Distances Checkpoint Distance (km) Accumulated Distance (km) Difficulty Start to CP1 15.9 15.9 CP1 to CP2 8.9 24.8 CP2 to CP3 9.9 34.7 CP3 to CP4 12.7 47.4 CP4 to CP5 7.7 55.1 CP5 to CP6 5.9 61.0 CP6 to CP7 8.5 69.5 CP7 to CP8 8.9 78.4 CP8 to CP9 9.5 87.9 CP9 to Finish 12.1 100 CP Elevation Height (m) Tai Mo Shan 1000 800 Ma On Shan Beacon Hill 600 Tate’s Cairn Kai Kung Shan Needle Hill 400 200 Start 10 20 30 40 50 60 70 80 90 Finish CP1 CP2 CP3 CP4 CP5 CP6 CP7 CP8 CP9 Distance (km)(Km) 29 CHECKPOINTS (CPS) All teams must register at a CP at or before its closing time in order to be allowed to proceed. -

Hong Kong Multisports Association 香 港 全 能
HONG KONG MULTISPORTS ASSOCIATION 香港全能運動學會 Ramp Challenge 2014 2nd Station Tsuen Wan- Tai Mo Shan Event Information Organizer: HK Multisports Association Objectives: to promote cross-country/trail running; enhance friendship and teamwork Receiving Charity: Hong Kong Island Private Hospitals Chaplaincy Committee Date :07-12-2013 (SUN) Start Time: 0900 Venue:Tai Mo Shan country Park Family Walk (Car Park) https://goo.gl/maps/0XuLA Registration time:0745-0845 Transportation: Tai Mo Shan Family Walk (Car Park) Take bus no. 51 in Tsuen Wan and get off at the Tsuen Kam Country Park Management Centre. From there, it is a 3-minute walk to the Rotary Club Park. http://www.kmb.hk/en/services/search.html Start point:Will be set up next to the Tai Mo Shan Road (Car Park) Distance:12KM Time Limit:2 Hrs Race route:Along Tai Mo Shan Road, straight on top, after a restricted area of the gate, continue straight on hill top sites, along tai mo shan forest trail Pagoda return back, will be go in to the trail when back down hill to Finish point. Attention: Peak sections will be run by the twists and turns of the carriageway, there are many trails to shortcut, For contestants security purposes, is strictly prohibited ran trails sections, the discoverer will be disqualified! The race course includes a few sections of uphill and Maclehose trail/ Trails downhill step, During wet weather, these can be quite slippery. Please be careful and pay extra attention to others’ and your own safety. Number Bib: The bib must be visible in the front at all times during race and it is not transferable. -

TNF 100 and TNF 50 Hong Kong Route Descriptions
2017 TNF 100 and TNF 50 HONG KONG ROUTE DESCRIPTIONS Both the 50 km and 100 km races start at 8 AM on Saturday, December 16th inside the Tai Mei Tuk car park in the Northern New Territories of Hong Kong SAR. Baggage check will take place in The Hong Kong Federation of Youth Groups Tai Mei Tuk Outdoor Activities Center (HKFYG Center). Race assembly will take place on the bicycle path near Bradbury Jockey Club Youth Hostel, Hong Kong Youth Hostels Association. TNF 100 and TNF 50 Section 1: Tai Mei Tuk / Pat Sing Leng Nature Trail / Wu Kau Tang Approximate distance ~ 6.6 km Total elevation gain ~ 460 meters Total elevation loss ~ 356 meters Racers will run out the bicycle path and turn right onto Ting Kok Road for approximately 1 km. The course by pass the Plover Cove Visitor Center, Agriculture, Fisheries and Conservation Department (AFCD) onto Brides Pool Road for about 500 meters then meet a trail head before a Barbecue Site managed by AFCD. Take sharp left turn, run on stone step trail all the way up until a junction with the Pat Sing Leng Nature Trail then turn right. The course continues along a footpath approximately 1.5 meters wide and gains altitude steadily. There are occasional breaks in the forest that afford views to the right of Plover Cove Reservoir. After attaining a height of land at about 350 meters, the path descends to an intersection where the Wilson Trail splits off to the left and racers continue on the Pat Sin Leng Nature Trail to the right. -

Survey Findings on Japanese World War II Military Installations in Hong Kong 78-94 by Lawrence W.C
THE HONG KONG INSTITUTE OF SURVEYORS Special issue to commemorate the 70th Anniversary of the Battle of Hong Kong, 8 December to 25 December 1941 InformatIon All rights reserved. No part of this Journal may be reproduced or transmitted in any form or by any means without the permission of the Hong Kong Institute of Surveyors. Contents of the Journal do not necessarily reflect the views or opinion of the Institute and no liability is accepted in relation thereto. Copyright © 2011 The Hong Kong Institute of Surveyors ISSN 1816-9554 Suite 801, 8/F, Jardine House, 1 Connaught Place, Central, Hong Kong Telephone 電話:(852) 2526 3679 Fax 傳真:(852) 2868 4612 Website 網址:www.hkis.org.hk Email 電郵:[email protected] EdItorIal Board Honorary Editor Dr K K Lo Gilbert Kwok Department of Building and Real Estate The Hong Kong Institute of Surveyors The Hong Kong Polytechnic University Hong Kong SAR, People’s Republic of China Hong Kong SAR, People’s Republic of China Chairman and Editor-in-Chief Dr King-fai Man Professor Kwong-wing Chau Department of Building and Real Estate Department of Real Estate and Construction The Hong Kong Polytechnic University The University of Hong Kong Hong Kong SAR, People’s Republic of China Hong Kong SAR, People’s Republic of China Professor Esmond Mok Editor Vol 21 Issue 2 Department of Land Surveying and Geo-Informatics Professor Lawrence Lai The Hong Kong Polytechnic University Department of Real Estate and Construction Hong Kong SAR, People’s Republic of China The University of Hong Kong Hong Kong SAR, People’s Republic -

The North Face 100® Hong Kong
The North Face 100® Hong Kong TNF 100 and TNF 50 Hong Kong Route Descriptions: Both the 50 km and 100 km races start at 8 AM on Saturday, December 12th inside the Tai Mei Tuk car park in the Northern New Territories of Hong Kong SAR. Baggage check will take place by the Bradbury Camp Youth Hostel. Race assembly will take place on the bicycle path near Bradbury Camp. TNF 100 and TNF 50 Section 1: Tai Mei Tuk/Pat Sing Leng Nature Trail/Wu Kau Tang Approximate distance ~ 6.6 km Total elevation gain ~ 400 meters Total elevation loss ~ 315 meters Racers will run out the bicycle path and turn right onto Bride’s Pool Road for approximately 1 km. The course turns left by the AFCD/Plover Cove Visitor Center and onto Pat Sing Leng Nature Trail. The course continues along a footpath approximately 1.5 meters wide and gains altitude steadily. There are occasional breaks in the forest that afford views to the right of Plover Cove Reservoir. After attaining a height of land at about 300 meters, the path descends to an intersection where the Wilson Trail splits off to the left and racers continue on the Pat Sin Leng Nature Trail to the right. Following a descent, a stream crossing, a short climb and steeper descent on stone stairs, the course arrives at Bride’s Pool Road. Although the race organizer and the police will manage traffic control, racers should cross the road with caution. The road is also popular with cyclists who tend to disregard instructions to give way to runners crossing the road. -

District : Tsuen
District : Tsuen Wan Proposed District Council Constituency Areas +/- % of Population Projected Quota Code Proposed Name Boundary Description Major Estates/Areas Population (16 599) K01 Tak Wah 15 475 -6.77 N Sai Lau Kok Road 1. CHUNG ON BUILDING 2. FOU WAH CENTRE NE Sai Lau Kok Road 3. HO FAI GARDEN E Kwan Mun Hau Street, Sai Lau Kok Road 4. KOLOUR - TSUEN WAN 1 5. TAK YAN BUILDING (PART) : Shing Mun Road Stage 2 SE Kwan Mun Hau Street Stage 4 S Sha Tsui Road Stage 6 Stage 7 SW Sha Tsui Road Stage 8 W Sha Tsui Road, Tai Ho Road 6. TSUEN CHEONG CENTRE 7. TSUEN WAN TOWN SQUARE NW Tai Ho Road, Tai Ho Road North 8. WAH SHING BUILDING K 1 District : Tsuen Wan Proposed District Council Constituency Areas +/- % of Population Projected Quota Code Proposed Name Boundary Description Major Estates/Areas Population (16 599) K02 Yeung Uk Road 17 799 +7.23 N Kwan Mun Hau Street, Sha Tsui Road 1. BO SHEK MANSION 2. EAST ASIA GARDENS NE Castle Peak Road - Tsuen Wan 3. HARMONY GARDEN Texaco Interchange, Texaco Road 4. NEW HAVEN 5. SHEUNG CHUI COURT Texaco Road Flyover 6. TSUEN WAN GARDEN E Texaco Road 7. WEALTHY GARDEN SE Texaco Road S Yeung Uk Road SW Yeung Uk Road W Chung On Street, Ho Pui Street Yeung Uk Road NW Chuen Lung Street, Sha Tsui Road K 2 District : Tsuen Wan Proposed District Council Constituency Areas +/- % of Population Projected Quota Code Proposed Name Boundary Description Major Estates/Areas Population (16 599) K03 Tsuen Wan South 19 623 +18.22 N Chuen Lung Street, Ho Pui Street 1.