Remi-Seq Handbook
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Livingston County Guide To
LIVINGSTON COUNTY GUIDE TO RE-USE RE-DUCE RE-CYCLE & SAFE DISPOSAL This guide was developed to assist residents and businesses of Livingston County dispose of unwanted items in an environmentally friendly manner. This guide was designed for informational purposes only. This guide is not an endorsement of the businesses mentioned herein. It is a good practice to always call first to confirm the business or organizations’ services. Livingston County Solid Waste Program 2300 E. Grand River, Suite 105 Howell, MI 48843-7581 Phone: 517-545-9609 www.livgov.com/dpw E-mail: [email protected] Revised 2-2015 TABLE OF CONTENTS Page/s AMMUNITION & EXPLOSIVES 3 APPLIANCES & LARGE ITEM DISPOSAL 3 ARTS & CRAFTS SUPPLIES (including recycling Christmas lights) 4 ASBESTOS 4 AUDIO – See: CDs, Disks, Video Tapes, Albums, Records 4 AUTOMOBILES 5 AUTOMOTIVE FLUIDS 6 BATTERIES – All types (and locations of the drop-off buckets) 7, 8 BUILDING MATERIALS & CONSTRUCTION DEBRIS 7 CDs – Disks, Tapes, Video Tapes, Albums, Records & DVDs 9 CELLULAR PHONES 9 CLOTHING & SHOES 9 COMPUTERS & ELECTRONICS 10 DUMPS (See Landfills) 10 EYEGLASSES & HEARING AIDS 10 FIRE EXTINGUISHERS 10 FLUORESCENT BULBS AND TUBES 10 FOOD ITEMS 11 FURNITURE AND LARGE ITEMS – Donation or Disposal 11 HOUSEHOLD HAZARDOUS WASTE 11 LANDFILLS, DUMPS AND TRANSFER STATIONS 12 LAWN & GARDEN EQUIPMENT 12 MERCURY – Thermometers, Thermostats 12,18 METAL 13 NEWSPAPER 13 OFFICE SUPPLIES AND EQUIPMENT 13 PACKAGING or SHIPPING MATERIALS 14 PAINT – Latex & Oil-Based Paints 14 PAPER SHREDDING-Check with your Townships and/or Recycle Livingston (pg. 16) for events PESTICIDES / HERBICIDES (Also disposal of commercial containers) 14,15 PETS ITEMS AND SUPPLIES 15 PHARMACEUTICALS and Over-the-Counter Medicines Disposal 15 PLASTICS RECYCLING – Including plastic shopping bags 15 PRINTER CARTRIDGES 16 PROPANE TANKS 16 RECYCLING CENTERS 16 SHARPS – Needles, injectors, and lancets 17 SHREDDING – Contacting your township for their programs or Recycle Livingston (pg. -

UN3373 Sample Transport Are You at Risk?
® UN3373 Sample Transport Are You at Risk? Find out how to package and transport biological samples safely and in accordance with the UN3373 regulations. ■ Supporting Category B sample transport compliance ■ Transportation solutions for Road and Air ■ A range of options for Secondary and Outer packaging Contents Page Introduction 3 UnderstandingtheRegulations3 Secondary Packaging - SpeciSafe® Mailing Packs4-6 - Absorbent Sheets7 - 95kPa Pouches :ADR and IATA8 - 95kPa Pouches with Absorbent8 Outer Containers - Rigid Outer Containers9 - UN3373 Sample Carriers9 - UN3373 Labels and Security Seals9 - Mailing Bags and Envelopes6 CompleteMailingSystems7 2 Introduction Understanding the Regulations UN3373 Standard – Key Requirements Whether for medical diagnostic purposes, clinical trials or research, getting patient and other biologically hazardous Packaging must be strong and of good quality to withstand samples safely from their source to the laboratory can be vibration or changes in temperature, humidity or pressure problematic. encountered during transport. It must prevent any loss of contents. The P650 packaging instructions for liquid substances state:- i. The primary receptacle(s) shall be leak proof ii. The secondary packaging shall be leak proof c. If multiple fragile primary receptacles are placed in a single secondary packaging, they shall be either individually wrapped or separated to prevent contact between them d. Absorbent material shall be placed between the primary receptacle(s) and the secondary packaging. The absorbent material shall be in quantitysufficient to absorb the entire contents of the primary receptacle(s) so that any release of the liquid Biological specimens fall under the Dangerous Goods substance will not compromise the integrity of the Regulations since they may contain pathogens. Their cushioning material or of the outer packaging transport is strictly controlled and it is a legal and mandatory e. -

Front Cover 2 Postal Bulletin 22300 (12-16-10)
Front Cover 2 postal bulletin 22300 (12-16-10) Contents COVER STORY Retail Field Information Kit: January 2011 Price Changes. 3 Stop Sending Copies of PS Form 8176, Premium Forwarding Service Application, to Headquarters . 115 POLICIES, PROCEDURES, AND FORMS UPDATES Supply Management SmartPay Purchase Card Program Accountability Manuals Acknowledgement, Account Setup, and Maintenance DMM Revision: Domestic Shipping Services Pricing Documents Update . 115 and Mailing Standards Changes. 10 DMM Revision: 2011 Changes for Domestic Mailing Services . 34 DMM Revision: Products Mailable at Nonprofit PULL-OUT INFORMATION Standard Mail Prices. 40 Fraud DMM Revision: Clarification of the Post Office Withholding of Mail Order . 47 Box Lock Replacement Fee . 40 Invalid Express Mail Corporate Account Numbers . 49 DMM Revision: Optional Mail Preparation Standards Missing, Lost, or Stolen U.S. Money Order Forms . 51 for Flat-Size Mailpieces in FSS Zones . 41 Missing, Lost, or Stolen Canadian Money Order Forms . 56 DMM Revision: Extension of the Simplified Verifying U.S. Postal Service Money Orders . 59 Address Format. 46 Counterfeit Canadian Money Order Forms . 59 DMM Revision: Submission of Electronic Documentation Toll-Free Number Available to Verify Canadian With Comailed and Copalletized Mailings . 73 Money Orders . 59 DMM Revision: Open and Distribute — Express Mail Other Information and Priority Mail Changes and Updates . 76 Overseas Military/Diplomatic Mail . 60 DMM Revision: Labeling List Changes . 79 Missing Children Posters. 65 IMM Revision: Changes to Pricing and Mailing Thrift Savings Plan Fact Sheet. 69 Standards for International Shipping Services . 82 IMM Revision: Israel Is Changing to Price Group 5 for First-Class Mail International Service and Airmail M-bag Service. -
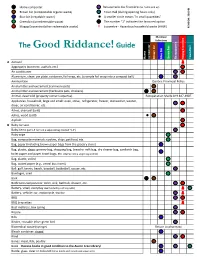
The Good Riddance!Guide
Home composter Ressourcerie des Frontières (at-home pick-up) Brown bin (compostable organic waste) Town Hall (during opening hours only) Blue bin (recyclable waste) A smaller circle means "in small quantities" Green bin (unredeemable waste) The number "1" indicates the favoured option Magog Ecocentre (other redeemable waste) Ecocentre - Hazardous household waste (HHW) REVISION: 2019-01 Municipal Collections The Good Riddance! Guide Home composter bin Brown bin Blue bin Green Ressourcerie Town Hall Magog Ecocentre Magog A Aerosol Aggregates (concrete, asphalt, etc.) Air conditioner Aluminium, clean: pie plate, container, foil wrap, etc. (crumple foil wrap into a compact ball) Ammunition Quebec Provincial Police Animal litter and excrement (carnivore pets) Animal litter and excrement (herbivore pets, chickens) Animal, dead wild (property owner's expense) Récupération Maillé 819 847-4907 Appliances, household, large and small: oven, stove, refrigerator, freezer, dishwasher, washer, dryer, air conditioner, etc. Ashes, charcoal (cold) Ashes, wood (cold) Asphalt B Baby car seat Baby items (put 0-5 items in a separate bag marked "0-5") Baby wipe Bag, composite materials: cookies, chips, pet food, etc. Bag, paper (including brown paper bags from the grocery store) Bag, plastic, clean: grocery bag, shopping bag, bread or milk bag, dry cleaner bag, sandwich bag, toilet paper and paper towel bags, etc. (Gather into a single bag and tie) Bag, plastic, soiled Bag, waxed paper (e.g., cereal box insert) Ball: golf, tennis, beach, baseball, basketball, -

Move-Out Procedures Dear Departing Residents
Move-Out Procedures Dear Departing Residents, As your move-out date approaches, Westside Village would like to take this opportunity to thank you for renting from us over the past year. In addition, we are providing you with this move-out guide/checklist for move-out procedures. Please read this entire packet carefully which provides you with very detailed and helpful information. This is how you get a security deposit refund! Westside Village prefers to return your entire security deposit. Use this information to your advantage so that you can receive your full security deposit back. Encountering apartments with damages, that are not properly cleaned, or that have personal belongings left behind will result in charges against your security deposit. In some circumstances, charges can also exceed the security deposit amount resulting in additional money that will be owed to Westside Village. By following these move-out guide instructions/procedures, you can leave your unit in the best possible condition. Leaving your apartment as outlined per the instructions in this move-out guide will save all parties involved time and money. This packet includes the following information for your reference: 1. Damaged Item Cost Sheet - In the event that anything was damaged in your apartment during the lease period, this list provides an estimated cost for replacement. Please note that these costs do not include sales tax and are only estimated costs, which may therefore vary depending on the nature and extent of the damage. 2. Security Deposit Check Return Form - As per your lease agreement you are required to submit a forwarding address so that we can return your security deposit check in a timely manner. -

Move-Out Procedures Dear Departing Residents
Move-Out Procedures Dear Departing Residents, As your move-out date approaches, Westside Village would like to take this opportunity to thank you for renting from us over the past year. In addition, we are providing you with this move-out guide/checklist for move-out procedures. Please read this entire packet carefully which provides you with very detailed and helpful information. This is how you get a security deposit refund! Westside Village prefers to return your entire security deposit. Use this information to your advantage so that you can receive your full security deposit back. Encountering apartments with damages, that are not properly cleaned, or that have personal belongings left behind will result in charges against your security deposit. In some circumstances, charges can also exceed the security deposit amount resulting in additional money that will be owed to Westside Village. By following these move-out guide instructions/procedures, you can leave your unit in the best possible condition. Leaving your apartment as outlined per the instructions in this move-out guide will save all parties involved time and money. This packet includes the following information for your reference: 1. Damaged Item Cost Sheet - In the event that anything was damaged in your apartment during the lease period, this list provides an estimated cost for replacement. Please note that these costs do not include sales tax and are only estimated costs, which may therefore vary depending on the nature and extent of the damage. 2. Cleaning Cost Sheet/Cleaning Guide - In the event that your apartment has not been cleaned and/or was not cleaned properly at move out, we will have the apartment cleaned and you will be charged accordingly. -

PACKING SLIP & SHIPPING INSTRUCTIONS Mail To: Arch Enterprises | 2501 Lakeview Road | Mexico, MO | 65265
PHONE: (573) 581-3110 FAX: (573) 581-3117 EMAIL: [email protected] WEB: www.ArchEnterprises.com PACKING SLIP & SHIPPING INSTRUCTIONS Mail To: Arch Enterprises | 2501 Lakeview Road | Mexico, MO | 65265 Customer Name: Shipping Date: Phone Number: Email Address: Mailing Address: Special Notes: 1. Place scrap in a plastic bag or other plastic container. 2. Place bag or container in a padded envelope or shipping container. 3. Make sure your shipment is secure; do not write gold, silver or platinum on your shipment. 4. Please ship items, USPS Priority Mail (preferred), DHL, UPS, or FedEx with a Delivery Signature Required. 5. Please include a copy of your packing slip and any email with your shipment. PLEASE CALL 1-800-835-0478 OR EMAIL US AT [email protected] WITH ANY QUESTIONS Turnaround Time: Our turnaround time for payment on jewelry, coins & bars is 21 business days. All other material, depending on the type, can range from 8 to 10 business weeks and there are certain types of material that may take additional time beyond the 10 business weeks to process payment on. Please inquire if you have questions. Disclaimer: Payment will depend on the actual weight and composition of material received. Please weigh all material and post on the packing list and include with your shipment. We will notify you if the material is not as described before processing. Materials we cannot process will be shipped back to the customer within 15 days at the customer’s expense. Items left over 15 days will not be returned. Arch recommends shipping your material via USPS Priority with a signature. -

Evidence Submission Guidelines
EVIDENCE SUBMISSION GUIDELINES US Fish and Wildlife Service National Fish and Wildlife Forensic Laboratory 1490 East Main Street Ashland, OR 97520 Introduction The Mission of the National Fish and Wildlife Forensics Laboratory (Laboratory) is to provide forensic support in wildlife investigations. The purpose of this guide is to help you with proper, and safe evidence documentation, packaging and shipping procedures so that your evidence shipment meets: 1) U.S. Department of Transportation regulations 2) Laboratory requirements for analysis 3) Requirements for presentation in courts of law Good Communication Call the Laboratory if you have any additional questions about how to best ship your evidence to the Lab. We are here to serve your needs. Contact us at: National Fish and Wildlife Forensics Laboratory 1490 East Main Street Ashland, OR 97520 Phone: (541) 482-4191 Fax: (541) 482-4989 Email: [email protected] * * * * OM-EU-1000 2/28/2013 Page 1 of 25 IMPORTANT NOTE on SHIPPING HAZARDOUS MATERIALS The U.S. Department of Transportation (USDOT) requires that Anyone who ships hazardous materials be trained and tested. 49 CFR Subpart H, Parts 172-700-704 What Documents are Needed for Submission? Furthermore, Anyone who knowingly violates a requirement of the Federal hazardous material transportation law is liable to civil penalties which may include fines and imprisonment. 49 CFR Part 171.1 (g) The types of hazardous materials encountered in shipping evidence include, but are not limited to: - Pesticides, poisons, and other chemicals -

USPS Priority Mail Box Size Guide
USPS® Priority Mail® Box Size Guide The table below shows options for different Priority Mail Boxes and Envelopes, along with dimensions, online postage pricing and weight options. PRIORITY MAIL BOXES Outside Outside Outside Online Box Image Description Box Box Box Postage Weight Packaging SKU Length Width Height Price Limit 10 pack Ships in O-BOX4 Non-Flat Rate Starts at Up to 7 1/4" 7 1/4" 6 1/2" packs of Small Cube Box $5.05 70 lbs. 10 or 25 25 pack O-BOX4X 10 pack Ships in O-BOX7 Non-Flat Rate Starts at Up to 12 1/4" 12 1/4" 8 1/2" packs of Large Cube Box $5.05 70 lbs. 10 or 25 25 pack O-BOX7X 10 pack Ships in 0-SHOEBOX Non-Flat Rate Starts at Up to 7 5/8" 5 1/4" 14 5/8" packs of Shoebox $5.05 70 lbs. 10 or 25 25 pack 0-SHOEBOX-X 10 pack Non-Flat Rate Ships in O-1097 Starts at Up to Small Box, 11 5/8" 2 1/2" 13 7/16" packs of $5.05 70 lbs. Side-Loading 10 or 25 25 pack O-1097X 10 pack Non-Flat Rate Ships in O-1092 Starts at Up to Medium Box, 12 1/4" 2 7/8" 13 11/16" packs of $5.05 70 lbs. Side-Loading 10 or 25 25 pack O-1092X Order USPS Supplies in Bulk! Call 1-800-610-8734. 10 pack Non-Flat Rate Ships in O-1095X Starts at Up to Small Box, 12 7/16" 3 1/8" 15 5/8" packs of $5.05 70 lbs. -
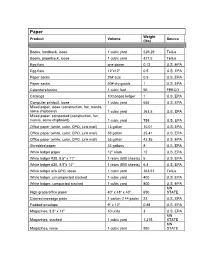
Product Volume to Weight Conversion Table
Paper Weight Product Volume Source (lbs) Books, hardback, loose 1 cubic yard 529.29 Tellus Books, paperback, loose 1 cubic yard 427.5 Tellus Egg flats one dozen 0.12 U.S. EPA Egg flats 12"x12" 0.5 U.S. EPA Paper sacks 25# size 0.5 U.S. EPA Paper sacks 50# dry goods 1 U.S. EPA Calendars/books 1 cubic foot 50 FEECO Catalogs 100 pages ledger 1 U.S. EPA Computer printout, loose 1 cubic yard 655 U.S. EPA Mixed paper, loose (construction, fax, manila, some chipboard) 1 cubic yard 363.5 U.S. EPA Mixed paper, compacted (construction, fax, manila, some chipboard) 1 cubic yard 755 U.S. EPA Office paper (white, color, CPO, junk mail) 13 gallon 10.01 U.S. EPA Office paper (white, color, CPO, junk mail) 33 gallon 25.41 U.S. EPA Office paper (white, color, CPO, junk mail) 55 gallon 42.35 U.S. EPA Shredded paper 33 gallons 8 U.S. EPA White ledger paper 12" stack 12 U.S. EPA White ledger #20, 8.5" x 11" 1 ream (500 sheets) 5 U.S. EPA White ledger #20, 8.5"x 14" 1 ream (500 sheets) 6.4 U.S. EPA White ledger w/o CPO, loose 1 cubic yard 363.51 Tellus White ledger, uncompacted stacked 1 cubic yard 400 U.S. EPA White ledger, compacted stacked 1 cubic yard 800 U.S. EPA MN High grade/office paper 40" x 48" x 40" 650 STATE Colored message pads 1 carton (144 pads) 22 U.S. -
Recycle Guide Is � Repair and Recondition Faulty Electronics Created to Help You Recycle Wisely at Appliances and Furniture to Extend Their Use- Ful Lives Guide Home
HISTORICAL CONTEXT PROMOTING THE « 3RV-E » Quebec enacted a law “Politique Québecoise Reduce at source : de gestion des matières résiduelles 1998- Buy lightly packaged goods Regional County Municipality 2008”. A proposed “Action Plan” for waste Buy items that have more than one use of Temiscamingue management to favour reduction at source for instead of disposable products. (to dispose after one use) a healthier environment. Buy product made from or packaged in recy- cled materials An objective of 60 % was set for each Buy in bulk instead of single serving Buy reusable and refillable containers municipality for the recycling of glass, Don’t buy things you don’t really need metal, plastic and cardboard. Reuse : Recycling must be an Consider the reuse of packaging (reuse plastic containers, metal box, plastic bag, integral part in every glass jars) Temiscamingue Convert scrap paper into memo pads Recycle citizen’s daily life. Choose reusable beer or pop bottles The recycle guide is Repair and recondition faulty electronics created to help you recycle wisely at appliances and furniture to extend their use- ful lives Guide home. Recycling : This guide will give you useful Take an active part in your community recy- suggestions based on the practice of cling program (paper, cardboard, glass, metal, plastic). the “3RV-E”: Reduce at source, reuse, recycle, valorization and elimination this should be the order followed Valorization : when doing residual recycling Leave the grass clipping on the ground. management. Home compost kitchen and garden waste. Elimination : Regional County Municipality of Recycle is a great way to take care of Only throw material with no reusable, Temiscamingue our environment. -

0 Philadelphia Police Department Directive 12.15
0 PHILADELPHIA POLICE DEPARTMENT DIRECTIVE 12.15 Issued Date: 03-13-15 Effective Date: 03-13-15 Updated Date: 06-15-20 SUBJECT: PROPERTY TAKEN INTO CUSTODY PLEAC 1.4.3, 1.4.4, 1.8.1, 3.6.1, 3.6.3, 3.6.4, 3.6.6 ______________________________________________________________________________ INDEX SECTION TITLE PAGE NO. 1 Policy 1 2 Property Classifications 1 3 Control and Issuance of Property Receipts (75-3) 3 4 Preparation of Property Receipts (75-3) 5 5 Storage Agencies and Limitations 7 6 Office of Forensic Science Evidence Packaging Guidelines 10 7 Transfer of Items to Storage 21 8 Evidence Intake Procedure 23 9 Cash Intake Procedure 27 10 Distribution of Property Receipts (75-3) 31 11 Obtaining Evidence for Court 33 12 Dispositions on Orders of the Court 34 13 Disposition of Property by Evidence Custodian 36 14 Audits and Inspections 38 15 Gun Buybacks 38 Appendix “A” Preparation of Property Receipts (75-3) Instructions Appendix “B” Example Memo – Withdrawal of Evidence without Property Receipt Appendix “C” Continuation of Property Receipt (75-3A) Appendix “D” Example Memo – Request for Disposal of Liquor PHILADELPHIA POLICE DEPARTMENT DIRECTIVE 12.15 Issued Date: 03-13-15 Effective Date: 03-13-15 Updated Date: 06-15-20 SUBJECT: PROPERTY TAKEN INTO CUSTODY PLEAC 1.4.3, 1.4.4, 1.8.1, 3.6.1, 3.6.3, 3.6.4, 3.6.6 ______________________________________________________________________________ 1. POLICY A. It is the policy of the Philadelphia Police Department that all property/money that is confiscated or collected will be recorded on a property receipt, thus initiating the chain of custody.