Self Injury in 1P36 Deletion Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phenotype Correlations in Individuals with Pathogenic RERE Variants
Genotype-phenotype correlations in individuals with pathogenic RERE variants Valerie K. Jordan,1 Brieana Fregeau,2 Xiaoyan Ge,3,4 Jessica Giordano,5 Ronald J. Wapner,5 Tugce B. Balci,6 Melissa T. Carter,6 John A. Bernat,7 Amanda N. Moccia,8 Anshika Srivastava,8 Donna M. Martin,8,9 Stephanie L. Bielas,8 John Pappas,10 Melissa D. Svoboda,11 Marlène Rio,12,13 Nathalie Boddaert,12,14 Vincent Cantagrel,12,15 Andrea M. Lewis,3,16 Fernando Scaglia,3,16 Undiagnosed Diseases Network, Jennefer N. Kohler,17 Jonathan A. Bernstein,17 Annika M. Dries,17 Jill A. Rosenfeld,3 Colette DeFilippo,18 Willa Thorson,19 Yaping Yang,3,4 Elliott H. Sherr,2 Weimin Bi,3,4 Daryl A. Scott1,3,16* 1) Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX, USA 2) Department of Neurology, University of California, San Francisco, San Francisco, CA, USA 3) Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA 4) Baylor Genetics, Houston, TX, USA 5) Institute of Genomic Medicine and Department of OB/GYN, Columbia University Medical Center, New York, NY, USA 6) Department of Genetics, Children’s Hospital of Eastern Ontario, Ottawa, ON, Canada 7) Stead Family Department of Pediatrics, The University of Iowa, Iowa City, IA, USA 8) Department of Human Genetics, University of Michigan Medical School, Ann Arbor, MI, USA This is the author manuscript accepted for publication and has undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. -

The Advantage of Genome-Wide Microarrays Over Targeted Approaches
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/70828 Please be advised that this information was generated on 2021-09-24 and may be subject to change. COPY NUMBER VARIATION AND MENTAL RETARDATION opmaak koolen.indd 1 10-09-2008 10:11:31 Copy number variation and mental retardation The studies presented in this thesis were performed at the Department of Human Genetics, Radboud University Nijmegen Medical Center, Nijmegen, the Netherlands. The research was supported by a grant from the Netherlands Organization for Health Research and Development (ZonMw). Publication of this thesis was financially supported by the Department of Human Genetics, Radboud University Nijmegen Medical Center, Nijmegen, the Netherlands. ISBN/EAN 978-90-6464-290-6 © 2008 D.A. Koolen All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, by print or otherwise, without permission in writing from the author. Cover photo: Printed by: Ponsen & Looijen B.V., Wageningen opmaak koolen.indd 2 10-09-2008 10:11:31 Copy number variation and mental retardation Een wetenschappelijke proeve op het gebied van de Medische Wetenschappen Proefschrift ter verkrijging van de graad doctor aan de Radboud Universiteit Nijmegen op gezag van de rector magnificus prof. mr. S.C.J.J. Kortmann, volgens besluit van het College van Decanen in het openbaar te verdedigen op donderdag 6 november 2008 om 15.30 uur precies door David Aljosja Koolen geboren op 22 juni 1976 te ‘s-Gravenhage opmaak koolen.indd 3 10-09-2008 10:11:32 Promotor: Prof. -

RARE CHROMOSOME DISORDERS the Term, ‘Rare Chromosome Disorders’, Refers to Conditions Which
INFORMATION SHEET Page 1 COMPLEX LEARNING DIFFICULTIES AND DISABILITIES RESEARCH PROJECT (CLDD) RARE CHROMOSOME DISORDERS The term, ‘rare chromosome disorders’, refers to conditions which: 1. occur due to missing, duplicated or re-arranged chromosome material 2. have a low prevalence rate (thus not including chromosomal disorders such as Down syndrome). Chromosomes are structures found in the nuclei of cells in human bodies. Each chromosome contains thousands of genes which determine how we grow and develop. A typically developing person will have 23 pairs of chromosomes with one member of each pair being inherited from each parent, giving a total of 46 individual chromosomes. Two of these are the sex chromosomes which determine whether we are female (XX) or male (XY). The remaining 44 chromosomes are grouped in 22 pairs, numbered 1 to 22. The arms of a chromosome are called ‘p’ (shorter arm) and ‘q’ (long arm) (see Figure 1); these arms are separated into numerical regions, which in turn are divided into bands and sub-bands. p q Figure 1. Diagram of a chromosome Individually, rare chromosome disorders are extremely uncommon, with some being actually unique; however, collectively rare chromosome disorders make up at least one in every 200 live births, with babies either having symptoms from birth or early childhood, or being carriers of a chromosomal abnormality and experiencing the effects when they try to reproduce in later life (Searle and Hultén, 2009). Recent advances in technology and medical expertise has meant that chromosomes can be viewed at ever increasing magnifications, which is resulting in the detection of more complex defects. -

1P36 Deletion Syndrome: an Update
References (1) Rosenfeld JA, et al. Refinement of causative genes in YOU monosomy 1p36 through clinical and molecular cytogenetic characterization of small interstitial deletions. Am J Med CONTACT US Genet 2010, 152A:1951–1959. Chromosome Disorder Outreach (2) Jordan VK, et al. 1p36 deletion syndrome: an update. ARE Applications Clin Genet 2015, 8:189-200. P.O. Box 724 (3) Oiglane-Shlik E, et al. Monosomy 1p36 - A multifaceted Boca Raton, FL 33429-0724 and still enigmatic syndrome: Four clinically diverse cases with shared white matter abnormalities. Eur J Paed Neurol NOT 2014, 18:338-346. Family Helpline 561.395.4252 [email protected] (4) Battaglia A, et al. Further delineation of deletion 1p36 syndrome in 60 patients: A recognizable phenotype and common cause of developmental delay and mental www.chromodisorder.org ALONE retardation. Pediatrics 2008, 121:404-410. (5) Chan YTP, et al. Answer to "Clinical Quiz". HK J Paediatr (New Series) 2015, 20:212-214. Chromosome (6) Arndt A-K, et al. Fine mapping of the 1p36 deletion Disorder Outreach syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet 2013, 93: 67-77. (7) Zaveri HP, et al. Identification of critical regions and ABOUT US candidate genes for cardiovascular malformations and cardiomyopathy associated with deletions of chromosome 1p36 Deletion 1p36. PLOS ONE 2014, 9:e85600. Chromosome Disorder Outreach Syndrome Author: Colleen Donnelly provides support and information to anyone diagnosed with a rare chromosome change, (Monosomy 1p36) rearrangement or disorder. CDO actively promotes research and a positive community understanding of all chromosome disorders. CDO is a 501c3 organization founded in 1992. -

Type II Diabetes and Impaired Glucose Tolerance Due to Severe
Stagi et al. BMC Medical Genetics 2014, 15:16 http://www.biomedcentral.com/1471-2350/15/16 CASE REPORT Open Access Type II diabetes and impaired glucose tolerance due to severe hyperinsulinism in patients with 1p36 deletion syndrome and a Prader-Willi-like phenotype Stefano Stagi1,4*, Elisabetta Lapi2, Marilena Pantaleo2, Francesco Chiarelli3, Salvatore Seminara1 and Maurizio de Martino1 Abstract Background: Deletion of the subtelomeric region of 1p36 is one of the most common subtelomeric deletion syndromes. In monosomy 1p36, the presence of obesity is poorly defined, and glucose metabolism deficiency is rarely reported. However, the presence of a typical Prader-Willi-like phenotype in patients with monosomy 1p36 is controversial. Case presentation: In this report, we describe two female patients, one who is 6 years 2 months of age and another whois10years1monthofage,bothreferredtoourhospitalforobesity and a Prader-Willi-like phenotype. These patients presented with severe obesity (body mass index [BMI] was 26.4 and 27.7, respectively), hyperphagia and developmental delay. Analysis of basal hormone levels showed normal thyroid function and adrenal function but considerable basal hyperinsulinism (the insulin levels were 54.5 and 49.2 μU/ml, respectively). In patient 1, glycaemia was 75 mg/dl (HOMA-R 10.09), and the HbA1c level was 6.1%; in patient 2, glycaemia was 122 mg/dl, and the HbA1c level was 6.6% (HOMA-R 14.82). An oral glucose tolerance test demonstratedimpairedglucosetoleranceanddiabetes mellitus with marked insulin resistance (the peak insulin level for each patient was 197 and 279 μU/mL, respectively, while the 120’ insulin level of each patient was 167 and 234 μU/mL, respectively). -

Inside This Issue
Winter 2014 No. 77 Inside this issue Group News | Fundraising | Members’ Letters | One Family Living with Two Different Chromosome Disorders | Bristol Conference 2014 | Unique Leaflets | Christmas Card Order Form Sophie, Unique’s Chair of Trustees Dear Members, In the past month a few things have reminded me of why it is so important to make connections through Unique but also to draw support from other parents around us. I’ve just returned from Unique’s most recent family conference in Bristol where 150 of us parents and carers had a lovely time in workshops, meals and activities, chatting and watching our children milling around together like one big family since – although we had never met before – we have shared so many experiences in common. However in contrast I have also just met a new mum who has just moved to my area from far away with two toddlers, one with a rare joys of the internet, it is becoming easier to meet others with similar, chromosome disorder, who is starting from scratch with no even very rare, chromosome disorders around the world and to find professional, medical or social support. She reminds me of how yourself talking to them in the middle of the night about some lonely I felt when Max was newly diagnosed, when I knew no one interesting things our children share in common (obsession with with a disabled child let alone anyone with a rare chromosome catalogues, anyone?) And of course we also have an enormous disorder. Elsewhere our latest Unique Facebook group, Unique amount in common with so many parents of children with other Russia, is also just starting up – so far it includes just a small special needs or disabilities around us in our own communities who number of members sharing very different experiences to mine here will often be walking the same path as us. -

The Most Complete Solution on the Market About Us Our
NIPTNIPT NIPT–GeneSGKit ® Sistemas Genómicos has developed NIPT-GeneSGKit® Advanced, the most complete test available for noninvasive prenatal screening for chromosomal aneuploidies present in the fetus, as well as establishing gender. THE MOST COMPLETE SOLUTION ON THE MARKET NIPT-GeneSGKit® Advanced is CE-IVD* marked and contains reagents to process 12-48 samples with the corresponding bioinformatics analysis, visualization and personalized report by the GeneSystems© platform. The work process involves a “hands-out” Validated complete bioinformatic manipulation time, completed in 3 hours, analysis. once the circulating free DNA (cfDNA) has been obtained (5ng). Simple and immediate generation of Integration in user’s facilities. individualized reports. Sistemas Genómicos is a pioneering company in the application of Next ABOUT Generation Sequencing technology for genetic diagnosis. Accredited and certified by the major entities in quality assurance (ISO9001, ISO13485, ISO17025 ISO15189 and CLIA), we offer solutions to accelerate and standardize clinical genetic testing, US enabling clinicians to provide accurate diagnostics to their patients. GeneSGKits ® library OUR MISSION preparation We strive to provide the best diagnostic solutions with the best technology and knowledge available to maximize Sample sequencing on patient benefit. Illumina ® platforms Our NIPT allows the implementation of this service in hospitals and other centers’ facilities without externalizing any sample, with highly reliable results, reducing times for obtaining the report. Analysis and data interpretation on GeneSystems © *Chromosome 21 excluded 1 NIPT-GeneSGKit ® - Trisomy 21* associated with Down’s syndrome. NIPT-GeneSGKit® Advanced is an integrated solution - Trisomy 18 associated with Edwards’ syndrome. that includes bioinformatics analysis, visualization and - Trisomy 13 associated with Patau syndrome. -

HIGH RISK 1P36 This Pregnancy Is Classified As HIGH RISK by This Screen for a Deletion at 1P36, Which Is Associated with 1P36 Deletion Syndrome
Patient Report |FINAL Client: Example Client ABC123 Patient: Patient, Example 123 Test Drive Salt Lake City, UT 84108 DOB 4/3/1982 UNITED STATES Gender: Female Patient Identifiers: 01234567890ABCD, 012345 Physician: Doctor, Example Visit Number (FIN): 01234567890ABCD Collection Date: 01/01/2017 12:34 Non-Invasive Prenatal Testing for Fetal Aneuploidy with Microdeletions ARUP test code 2010232 Result Summary HIGH RISK 1p36 This pregnancy is classified as HIGH RISK by this screen for a deletion at 1p36, which is associated with 1p36 deletion syndrome. This result should be confirmed by a diagnostic test. Dependent on fetal fraction, 7 to 17% of pregnancies classified as HIGH RISK are found to have 1p36 deletion syndrome. TEST INFORMATION: Non-Invasive Prenatal Testing for Fetal Aneuploidy (Powered by Constellation) with or without Microdeletions METHODOLOGY: DNA isolated from the maternal blood, which contains placental DNA, is amplified at 13,300+ loci using a targeted PCR assay and sequenced using a high-throughput sequencer. Sequence data are analyzed using Natera's Constellation software to estimate the fetal copy number and identify whole chromosome abnormalities for chromosomes 13, 18, 21, X, and Y as well as fetal sex. Barring QC failures and fetal fractions below the performance limits of the algorithm, the minimum confidence threshold is 0.98 for a high risk call. For both low risk and high risk calls, the majority of specimens will have a confidence of >0.99 across all regions tested. If a sample fails to meet the quality threshold, no result will be reported for one or more chromosomes. Microdeletions: An additional 6,600+ loci are amplified to estimate the fetal copy numbers of chromosomal regions attributed to 22q11.2, Prader-Willi, Angelman, Cri-du-chat, and 1p36 deletion syndromes. -

Panorama™ Non-Invasive Prenatal Screening for Microdeletion Syndromes
NON-INVASIVE PRENATAL SCREENING FOR MICRODELETION SYNDROMES HALL 1 PANORAMA™ NON-INVASIVE PRENATAL SCREENING FOR MICRODELETION SYNDROMES MEGAN P. HALL, PH.D. INTRODUCTION PanoramaTM is a non-invasive prenatal screening test for fetal chromosomal anomalies. The screening test analyzes fetal cell-free DNA (cfDNA) isolated from maternal plasma and can be performed as early as 9 weeks of gestation with high accuracy. Panorama was originally designed to screen for Trisomy 21 (Down syndrome), Trisomy 18 (Edwards syndrome), Trisomy 13 (Patau syndrome), Monosomy X (Turner syndrome), sex chromosome trisomies, triploidy and, if requested, fetal sex (1-5). Recently, the clinical scope of Panorama was expanded to include screening for five microdeletion syndromes. This Panorama Extended Panel now screens for the 22q11.2 deletion (DiGeorge), 1p36 deletion, Cri-du-chat, Prader-Willi, and Angelman deletions. Validation studies have demonstrated sensitivities of greater than 93% and specificities of greater than 99% for each microdeletion condition. MICRODELETION PREVALENCE AND TRADITIONAL MICRODELETION DETECTION Clinically relevant microdeletions and microduplications are more common than previously thought, occurring in up to 1 in 60 pregnancies, and can occur in pregnancies lacking ultrasound anomalies (6). The combined at-birth incidence of the 5 microdeletion syndromes covered by this screening test is approximately 1 in 1,000 (7-11), approaching the overall rate observed for Down syndrome (12). The most common microdeletion, 22q11.2 deletion, is more common than Edwards syndrome and Patau syndrome combined, and is more common than cystic fibrosis (Figure 1)(10,12,13). Further, the risk for microdeletions is independent of maternal age, unlike whole chromosome aneuploidies like Trisomy 21 (Down syndrome) that are more prevalent in women of advanced maternal age. -

1P36 Microdeletion Syndrome: a Case Report
Acta Medica 2014; 3: 26–28 acta medica CASE REPORT 1p36 Microdeletion Syndrome: A Case Report Pınar Zengin AKKUŞ1, [MD] ABSTRACT Yavuz ŞAHİN2, [MD] 1p36 deletion syndrome is one of the most common microdeletion syndromes with an estimated prevalence of 1 in 5000. A 3-year-old girl with intellectu- Eda UTINE1, [MD] al disability and facial anomalies was admitted to the Genetics Outpatient 1 Koray BODUROĞLU , [MD] Clinic. The patient was born to consanguineous parents. She had seizures and growth retardation, behavioral problems including aggression and self-injurious behavior. On physical examination, she had low body weight, short stature, and dysmorphic facial characteristics including microceph- aly, prominent forehead, deep set eyes, straight eyebrows and micrognath- ia. Ophthalmologic, auditory and cardiac examinations were normal. Facial 1 Department of Pediatrics, Department of dysmorphic features and intellectual disability suggested presence of 1p36 Pediatric Genetics, microdeletion syndrome, and this was confirmed by karyotype analysis and 2 Department of Medical Genetics at Hacettepe fluorescence in situ hybridization (FISH): 46,XX,del (1) (p36.3) (CDC2L1- University, Faculty of Medicine ,CEB108-). The condition is caused by a deletion with variable breakpoints at * Corresponding Author: Pınar Zengin Akkuş, the distal tip of the short arm of chromosome. The deletion may at times be MD, Hacettepe University Faculty of Medicine, detected by high resolution karyotype, but mostly, FISH analysis is required Department of Pediatrics, Department of for definitive diagnosis. Pediatric Genetics, Sihhiye, 06100 Ankara, Turkey Key words: 1p36 microdeletion, intellectual disability, dysmorphic features e-mail [email protected] Received 31 March 2014; accepted 11 April 2014; pub- lished online 28 April 2014 Introduction 1p36 microdeletion syndrome is one of the most of 3 months. -

Non-Classical 1P36 Deletion in a Patient
Yokoyama et al. Mol Cytogenet (2020) 13:42 https://doi.org/10.1186/s13039-020-00510-5 CASE REPORT Open Access Non-classical 1p36 deletion in a patient with Duane retraction syndrome: case report and literature review Emiy Yokoyama1, Camilo E. Villarroel1, Sinhué Diaz2, Victoria Del Castillo1, Patricia Pérez‑Vera3, Consuelo Salas3, Samuel Gómez4, Reneé Barreda1, Bertha Molina5 and Sara Frias5,6* Abstract Background: Monosomy of 1p36 is considered the most common terminal microdeletion syndrome. It is character‑ ized by intellectual disability, growth retardation, seizures, congenital anomalies, and distinctive facial features that are absent when the deletion is proximal, beyond the 1p36.32 region. In patients with proximal deletions, little is known about the associated phenotype, since only a few cases have been reported in the literature. Ocular manifestations in patients with classical 1p36 monosomy are frequent and include strabismus, myopia, hypermetropia, and nystagmus. However, as of today only one patient with 1p36 deletion and Duane retraction syndrome (DRS) has been reported. Case presentation: We describe a patient with intellectual disability, facial dysmorphism, and bilateral Duane retrac‑ tion syndrome (DRS) type 1. Array CGH showed a 7.2 Mb de novo deletion from 1p36.31 to 1p36.21. Discussion: Our patient displayed DRS, which is not part of the classical phenotype and is not a common clinical feature in 1p36 deletion syndrome; we hypothesized that this could be associated with the overlapping deletion between the distal and proximal 1p36 regions. DRS is one of the Congenital Cranial Dysinnervation Disorders, and a genetic basis for the syndrome has been extensively reported. -
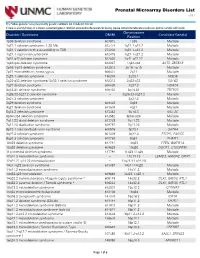
Prenatal Microarray Disorders List V19.1
Prenatal Microarray Disorders List v19.1 This "whole genome" array may identify genetic conditions not included in this list. If there is a family history of a known suspected genetic condition unrelated to the reason for testing, please contact the laboratory to discuss prior to sample submission. Chromosome Disorder / Syndrome OMIM Candidate Gene(s) Position 1p36 deletion syndrome 607872 1p36 Multiple 1q21.1 deletion syndrome, 1.35 Mb 612474 1q21.1-q21.2 Multiple 1q21.1 deletion with susceptibility to TAR 274000 1q21.1-q21.2 Multiple 1q21.1 duplication syndrome 612475 1q21.1-q21.2 Multiple 1q41-q42 deletion syndrome 612530 1q41-q42.12 Multiple 1q43-q44 deletion syndrome 612337 1q43-q44 AKT3, ZBTB18 2p16.1-p15 deletion syndrome 612513 2p16.1-p15 Multiple 2p21 microdeletion, homozygous 606407 2p21 Multiple 2q23.1 deletion syndrome 156200 2q23.1 MBD5 2q32-q33 deletion syndrome/ 2q33.1 deletion syndrome 612313 2q32-q33 SATB2 2q37 deletion syndrome 600430 2q37.3 HDAC4 3q13.31 deletion syndrome 615433 3q13.31 ZBTB20 3q26.33-3q27.2 deletion syndrome -- 3q26.33-3q27.2 Multiple 3q27.3 deletion syndrome -- 3q27.3 Multiple 3q29 deletion syndrome 609425 3q29 Multiple 4q21 deletion syndrome 613509 4q21 Multiple 5q14.3 deletion syndrome 613443 5q14.3 MEF2C 6pter-p24 deletion syndrome 612582 6pter-p24 Multiple 7q11.23 distal deletion syndrome 613729 7q11.23 Multiple 7q11.23 duplication syndrome 609757 7q11.23 Multiple 8p23.1 deletion/duplication syndrome 600576 8p23.1 GATA4 9q22.3 deletion syndrome 601309 9q22.3 PTCH1, FANCC 9q34.3 deletion syndrome