Syntrophic Butyrate and Propionate Oxidation Processes 491
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Core Sulphate-Reducing Microorganisms in Metal-Removing Semi-Passive Biochemical Reactors and the Co-Occurrence of Methanogens
microorganisms Article Core Sulphate-Reducing Microorganisms in Metal-Removing Semi-Passive Biochemical Reactors and the Co-Occurrence of Methanogens Maryam Rezadehbashi and Susan A. Baldwin * Chemical and Biological Engineering, University of British Columbia, 2360 East Mall, Vancouver, BC V6T 1Z3, Canada; [email protected] * Correspondence: [email protected]; Tel.: +1-604-822-1973 Received: 2 January 2018; Accepted: 17 February 2018; Published: 23 February 2018 Abstract: Biochemical reactors (BCRs) based on the stimulation of sulphate-reducing microorganisms (SRM) are emerging semi-passive remediation technologies for treatment of mine-influenced water. Their successful removal of metals and sulphate has been proven at the pilot-scale, but little is known about the types of SRM that grow in these systems and whether they are diverse or restricted to particular phylogenetic or taxonomic groups. A phylogenetic study of four established pilot-scale BCRs on three different mine sites compared the diversity of SRM growing in them. The mine sites were geographically distant from each other, nevertheless the BCRs selected for similar SRM types. Clostridia SRM related to Desulfosporosinus spp. known to be tolerant to high concentrations of copper were members of the core microbial community. Members of the SRM family Desulfobacteraceae were dominant, particularly those related to Desulfatirhabdium butyrativorans. Methanogens were dominant archaea and possibly were present at higher relative abundances than SRM in some BCRs. Both hydrogenotrophic and acetoclastic types were present. There were no strong negative or positive co-occurrence correlations of methanogen and SRM taxa. Knowing which SRM inhabit successfully operating BCRs allows practitioners to target these phylogenetic groups when selecting inoculum for future operations. -

Anaerobic Microbial LCFA Degradation in Bioreactors
Presented in Session PP3A – Bio-electrochemical Processes 11th IWA World Congress on Anaerobic Digestion 23-27 September 2007 Brisbane, Australia Anaerobic microbial LCFA degradation in bioreactors D.Z. Sousa*, M.A. Pereira*, J.I. Alves*, H. Smidt**, A.J.M. Stams**, M.M. Alves* * Institute for Biotechnology and Bioengineering, Center for Biological Engineering, University of Minho, Campus de Gualtar, 4710-057 Braga, Portugal (E-mail: [email protected]; [email protected]) ** Laboratory of Microbiology, Wageningen University, Hesselink van Suchtelenweg 4, 6703 CT Wageningen, The Netherlands Abstract This paper reviews recent results obtained on long-chain fatty acids (LCFA) anaerobic degradation. Two LCFA were used as model substrates: oleate, a mono-unsaturated LCFA, and palmitate, a saturated LCFA, both abundant in LCFA-rich wastewaters. 16S rRNA gene analysis of sludge samples submitted to continuous oleate- and palmitate-feeding followed by batch degradation of the accumulated LCFA demonstrated that bacterial communities were dominated by members of the Clostridiaceae and Syntrophomonadaceae families. Archaeal populations were mainly comprised of hydrogen-consuming microorganisms belonging to the genus Methanobacterium, and acetate- utilizers from the genera Methanosaeta and Methanosarcina. Enrichment cultures growing on oleate and palmitate, in the absence or presence of sulfate, gave more insight into the major players involved in the degradation of unsaturated and saturated LCFA. Syntrophomonas-related species were identified as predominant microorganisms in all the enrichment cultures. Microorganisms clustering within the family Syntrophobacteraceae were identified in the methanogenic and sulfate-reducing enrichments growing on palmitate. Distinct bacterial consortia were developed in oleate and palmitate enrichments, and observed differences might be related to the different degrees of saturation of these two LCFA. -

Assessment of Bacterial Species Present in Pasig River and Marikina River Soil Using 16S Rdna Phylogenetic Analysis
International Journal of Philippine Science and Technology, Vol. 08, No. 2, 2015 !73 SHORT COMMUNICATION Assessment of bacterial species present in Pasig River and Marikina River soil using 16S rDNA phylogenetic analysis Maria Constancia O. Carrillo*, Paul Kenny L. Ko, Arvin S. Marasigan, and Arlou Kristina J. Angeles Department of Physical Sciences and Mathematics, College of Arts and Sciences, University of the Philippines Manila, Padre Faura St., Ermita, Manila Philippines 1000 Abstract—The Pasig River system, which includes its major tributaries, the Marikina, Taguig-Pateros, and San Juan Rivers, is the most important river system in Metro Manila. It is known to be heavily polluted due to the dumping of domestic, industrial and solid wastes. Identification of microbial species present in the riverbed may be used to assess water and soil quality, and can help in assessing the river’s capability of supporting other flora and fauna. In this study, 16S rRNA gene or 16S rDNA sequences obtained from community bacterial DNA extracted from riverbed soil of Napindan (an upstream site along the Pasig River) and Vargas (which is along the Marikina River) were used to obtain a snapshot of the types of bacteria populating these sites. The 16S rDNA sequences of amplicons produced in PCR with total DNA extracted from soil samples as template were used to build clone libraries. Four positive clones were identified from each site and were sequenced. BLAST analysis revealed that none of the contiguous sequences obtained had complete sequence similarity to any known cultured bacterial species. Using the classification output of the Ribosomal Database Project (RDP) Classifier and DECIPHER programs, 16S rDNA sequences of closely related species were collated and used to construct a neighbor-joining phylogenetic tree using MEGA6. -

Crystalline Iron Oxides Stimulate Methanogenic Benzoate Degradation in Marine Sediment-Derived Enrichment Cultures
The ISME Journal https://doi.org/10.1038/s41396-020-00824-7 ARTICLE Crystalline iron oxides stimulate methanogenic benzoate degradation in marine sediment-derived enrichment cultures 1,2 1 3 1,2 1 David A. Aromokeye ● Oluwatobi E. Oni ● Jan Tebben ● Xiuran Yin ● Tim Richter-Heitmann ● 2,4 1 5 5 1 2,3 Jenny Wendt ● Rolf Nimzyk ● Sten Littmann ● Daniela Tienken ● Ajinkya C. Kulkarni ● Susann Henkel ● 2,4 2,4 1,3 2,3,4 1,2 Kai-Uwe Hinrichs ● Marcus Elvert ● Tilmann Harder ● Sabine Kasten ● Michael W. Friedrich Received: 19 May 2020 / Revised: 9 October 2020 / Accepted: 22 October 2020 © The Author(s) 2020. This article is published with open access Abstract Elevated dissolved iron concentrations in the methanic zone are typical geochemical signatures of rapidly accumulating marine sediments. These sediments are often characterized by co-burial of iron oxides with recalcitrant aromatic organic matter of terrigenous origin. Thus far, iron oxides are predicted to either impede organic matter degradation, aiding its preservation, or identified to enhance organic carbon oxidation via direct electron transfer. Here, we investigated the effect of various iron oxide phases with differing crystallinity (magnetite, hematite, and lepidocrocite) during microbial 1234567890();,: 1234567890();,: degradation of the aromatic model compound benzoate in methanic sediments. In slurry incubations with magnetite or hematite, concurrent iron reduction, and methanogenesis were stimulated during accelerated benzoate degradation with methanogenesis as the dominant electron sink. In contrast, with lepidocrocite, benzoate degradation, and methanogenesis were inhibited. These observations were reproducible in sediment-free enrichments, even after five successive transfers. Genes involved in the complete degradation of benzoate were identified in multiple metagenome assembled genomes. -
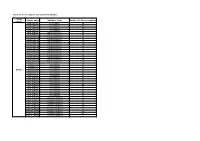
Table S1: List of Samples Included in the Analysis
Table S1: list of samples included in the analysis Study Sample name Inhibitory status Number of days at sampling number DNA.0P2T4 No inhibition 29 DNA.0P2T6 No inhibition 57 DNA.10P2T4 No inhibition 29 DNA.10P2T6 No inhibition 57 DNA.75P2T4 Phenol inhibition 29 DNA.75P2T6 Phenol inhibition 57 DNA.100P2T4 Phenol inhibition 29 DNA.100P2T6 Phenol inhibition 57 DNA.125P1T4 Phenol inhibition 29 DNA.125P1T6 Phenol inhibition 57 DNA.125P2T4 Phenol inhibition 29 DNA.125P2T6 Phenol inhibition 57 DNA.125P3T4 Phenol inhibition 29 DNA.125P3T6 Phenol inhibition 57 DNA.150P2T4 Phenol inhibition 29 DNA.150P2T6 Phenol inhibition 57 DNA.200P2T4 Phenol inhibition 29 DNA.200P2T6 Phenol inhibition 57 DNA.0N2T4 No inhibition 29 DNA.0N2T5 No inhibition 42 DNA.0N2T6 No inhibition 57 Study 1 DNA.5N2T4 No inhibition 29 DNA.5N2T5 No inhibition 42 DNA.5N2T6 No inhibition 57 DNA.10N2T4 No inhibition 29 DNA.10N2T5 No inhibition 42 DNA.10N2T6 No inhibition 57 DNA.15N2T4 No inhibition 29 DNA.15N2T5 No inhibition 42 DNA.15N2T6 No inhibition 57 DNA.25N2T4 No inhibition 29 DNA.25N2T5 No inhibition 42 DNA.25N2T6 No inhibition 57 DNA.75N2T4 Ammonia inhibition 29 DNA.75N2T5 Ammonia inhibition 42 DNA.75N2T6 Ammonia inhibition 57 DNA.100N2T4 Ammonia inhibition 29 DNA.100N2T5 Ammonia inhibition 42 DNA.100N2T6 Ammonia inhibition 57 DNA.250N2T4 Ammonia inhibition 29 DNA.250N2T5 Ammonia inhibition 42 DNA.250N2T6 Ammonia inhibition 57 nono2T3 No inhibition 16 noN2T4 Ammonia inhibition 23 noN2T8 Ammonia inhibition 60 noN2T9 Ammonia inhibition 85 noPhi2T4 Phenol inhibition 23 noPhi2T5 -

Dehalobacter Restrictus PER-K23T
Standards in Genomic Sciences (2013) 8:375-388 DOI:10.4056/sigs.3787426 Complete genome sequence of Dehalobacter restrictus T PER-K23 Thomas Kruse1*, Julien Maillard2, Lynne Goodwin3,4, Tanja Woyke3, Hazuki Teshima3,4, David Bruce3,4, Chris Detter3,4, Roxanne Tapia3,4, Cliff Han3,4, Marcel Huntemann3, Chia-Lin Wei3, James Han3, Amy Chen3, Nikos Kyrpides3, Ernest Szeto3, Victor Markowitz3, Natalia Ivanova3, Ioanna Pagani3, Amrita Pati3, Sam Pitluck3, Matt Nolan3, Christof Holliger2, and Hauke Smidt1 1 Wageningen University, Agrotechnology and Food Sciences, Laboratory of Microbiology, Dreijenplein 10, NL-6703 HB Wageningen, The Netherlands. 2 Ecole Polytechnique Fédérale de Lausanne (EPFL), School of Architecture, Civil and Environmental Engineering, Laboratory for Environmental Biotechnology, Station 6, CH- 1015 Lausanne, Switzerland. 3 DOE Joint Genome Institute, Walnut Creek, California, USA 4 Los Alamos National Laboratory, Bioscience Division, Los Alamos, New Mexico, USA *Corresponding author: Thomas Kruse ([email protected]) Keywords: Dehalobacter restrictus type strain, anaerobe, organohalide respiration, PCE, TCE, reductive dehalogenases Dehalobacter restrictus strain PER-K23 (DSM 9455) is the type strain of the species Dehalobacter restrictus. D. restrictus strain PER-K23 grows by organohalide respiration, cou- pling the oxidation of H2 to the reductive dechlorination of tetra- or trichloroethene. Growth has not been observed with any other electron donor or acceptor, nor has fermentative growth been shown. Here we introduce the first full genome of a pure culture within the ge- nus Dehalobacter. The 2,943,336 bp long genome contains 2,826 protein coding and 82 RNA genes, including 5 16S rRNA genes. Interestingly, the genome contains 25 predicted re- ductive dehalogenase genes, the majority of which appear to be full length. -

Tree Scale: 1 D Bacteria P Desulfobacterota C Jdfr-97 O Jdfr-97 F Jdfr-97 G Jdfr-97 S Jdfr-97 Sp002010915 WGS ID MTPG01
d Bacteria p Desulfobacterota c Thermodesulfobacteria o Thermodesulfobacteriales f Thermodesulfobacteriaceae g Thermodesulfobacterium s Thermodesulfobacterium commune WGS ID JQLF01 d Bacteria p Desulfobacterota c Thermodesulfobacteria o Thermodesulfobacteriales f Thermodesulfobacteriaceae g Thermosulfurimonas s Thermosulfurimonas dismutans WGS ID LWLG01 d Bacteria p Desulfobacterota c Desulfofervidia o Desulfofervidales f DG-60 g DG-60 s DG-60 sp001304365 WGS ID LJNA01 ID WGS sp001304365 DG-60 s DG-60 g DG-60 f Desulfofervidales o Desulfofervidia c Desulfobacterota p Bacteria d d Bacteria p Desulfobacterota c Desulfofervidia o Desulfofervidales f Desulfofervidaceae g Desulfofervidus s Desulfofervidus auxilii RS GCF 001577525 1 001577525 GCF RS auxilii Desulfofervidus s Desulfofervidus g Desulfofervidaceae f Desulfofervidales o Desulfofervidia c Desulfobacterota p Bacteria d d Bacteria p Desulfobacterota c Thermodesulfobacteria o Thermodesulfobacteriales f Thermodesulfatatoraceae g Thermodesulfatator s Thermodesulfatator atlanticus WGS ID ATXH01 d Bacteria p Desulfobacterota c Desulfobacteria o Desulfatiglandales f NaphS2 g 4484-190-2 s 4484-190-2 sp002050025 WGS ID MVDB01 ID WGS sp002050025 4484-190-2 s 4484-190-2 g NaphS2 f Desulfatiglandales o Desulfobacteria c Desulfobacterota p Bacteria d d Bacteria p Desulfobacterota c Thermodesulfobacteria o Thermodesulfobacteriales f Thermodesulfobacteriaceae g QOAM01 s QOAM01 sp003978075 WGS ID QOAM01 d Bacteria p Desulfobacterota c BSN033 o UBA8473 f UBA8473 g UBA8473 s UBA8473 sp002782605 WGS -

EXPERIMENTAL STUDIES on FERMENTATIVE FIRMICUTES from ANOXIC ENVIRONMENTS: ISOLATION, EVOLUTION, and THEIR GEOCHEMICAL IMPACTS By
EXPERIMENTAL STUDIES ON FERMENTATIVE FIRMICUTES FROM ANOXIC ENVIRONMENTS: ISOLATION, EVOLUTION, AND THEIR GEOCHEMICAL IMPACTS By JESSICA KEE EUN CHOI A dissertation submitted to the School of Graduate Studies Rutgers, The State University of New Jersey In partial fulfillment of the requirements For the degree of Doctor of Philosophy Graduate Program in Microbial Biology Written under the direction of Nathan Yee And approved by _______________________________________________________ _______________________________________________________ _______________________________________________________ _______________________________________________________ New Brunswick, New Jersey October 2017 ABSTRACT OF THE DISSERTATION Experimental studies on fermentative Firmicutes from anoxic environments: isolation, evolution and their geochemical impacts by JESSICA KEE EUN CHOI Dissertation director: Nathan Yee Fermentative microorganisms from the bacterial phylum Firmicutes are quite ubiquitous in subsurface environments and play an important biogeochemical role. For instance, fermenters have the ability to take complex molecules and break them into simpler compounds that serve as growth substrates for other organisms. The research presented here focuses on two groups of fermentative Firmicutes, one from the genus Clostridium and the other from the class Negativicutes. Clostridium species are well-known fermenters. Laboratory studies done so far have also displayed the capability to reduce Fe(III), yet the mechanism of this activity has not been investigated -

Research Article Review Jmb
J. Microbiol. Biotechnol. (2017), 27(0), 1–7 https://doi.org/10.4014/jmb.1707.07027 Research Article Review jmb Methods 20,546 sequences and all the archaeal datasets were normalized to 21,154 sequences by the “sub.sample” Bioinformatics Analysis command. The filtered sequences were classified against The raw read1 and read2 datasets was demultiplexed by the SILVA 16S reference database (Release 119) using a trimming the barcode sequences with no more than 1 naïve Bayesian classifier built in Mothur with an 80% mismatch. Then the sequences with the same ID were confidence score [5]. Sequences passing through all the picked from the remaining read1 and read2 datasets by a filtration were also clustered into OTUs at 6% dissimilarity self-written python script. Bases with average quality score level. Then a “classify.otu” function was utilized to assign lower than 25 over a 25 bases sliding window were the phylogenetic information to each OTU. excluded and sequences which contained any ambiguous base or had a final length shorter than 200 bases were Reference abandoned using Sickle [1]. The paired reads were assembled into contigs and any contigs with an ambiguous 1. Joshi NA, FJ. 2011. Sickle: A sliding-window, adaptive, base, more than 8 homopolymeric bases and fewer than 10 quality-based trimming tool for FastQ files (Version 1.33) bp overlaps were culled. After that, the contigs were [Software]. further trimmed to get rid of the contigs that have more 2. Schloss PD. 2010. The Effects of Alignment Quality, than 1 forward primer mismatch and 2 reverse primer Distance Calculation Method, Sequence Filtering, and Region on the Analysis of 16S rRNA Gene-Based Studies. -

Microbial Processes in Oil Fields: Culprits, Problems, and Opportunities
Provided for non-commercial research and educational use only. Not for reproduction, distribution or commercial use. This chapter was originally published in the book Advances in Applied Microbiology, Vol 66, published by Elsevier, and the attached copy is provided by Elsevier for the author's benefit and for the benefit of the author's institution, for non-commercial research and educational use including without limitation use in instruction at your institution, sending it to specific colleagues who know you, and providing a copy to your institution’s administrator. All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution’s website or repository, are prohibited. For exceptions, permission may be sought for such use through Elsevier's permissions site at: http://www.elsevier.com/locate/permissionusematerial From: Noha Youssef, Mostafa S. Elshahed, and Michael J. McInerney, Microbial Processes in Oil Fields: Culprits, Problems, and Opportunities. In Allen I. Laskin, Sima Sariaslani, and Geoffrey M. Gadd, editors: Advances in Applied Microbiology, Vol 66, Burlington: Academic Press, 2009, pp. 141-251. ISBN: 978-0-12-374788-4 © Copyright 2009 Elsevier Inc. Academic Press. Author's personal copy CHAPTER 6 Microbial Processes in Oil Fields: Culprits, Problems, and Opportunities Noha Youssef, Mostafa S. Elshahed, and Michael J. McInerney1 Contents I. Introduction 142 II. Factors Governing Oil Recovery 144 III. Microbial Ecology of Oil Reservoirs 147 A. Origins of microorganisms recovered from oil reservoirs 147 B. Microorganisms isolated from oil reservoirs 148 C. Culture-independent analysis of microbial communities in oil reservoirs 155 IV. -

Propionic Acid Degradation by Syntrophic Bacteria During Anaerobic Biowaste Digestion Propionic Acid Degradation Propionic WIREŁŁO
KARLSRUHERBERICHTE ZUR INGENIEURBIOLOGIE . MONIKA FELCHNER-Z WIREŁŁO Propionic Acid Degradation by Syntrophic Bacteria During Anaerobic Biowaste Digestion Propionic Acid Degradation Propionic WIREŁŁO . MONIKA FELCHNER-Z 49 . Monika Felchner-Z wirełło Propionic Acid Degradation by Syntrophic Bacteria During Anaerobic Biowaste Digestion Karlsruher Berichte zur Ingenieurbiologie Band 49 Institut für Ingenieurbiologie und Biotechnologie des Abwassers Karlsruher Institut für Technologie Herausgeber: Prof. Dr. rer. nat. J. Winter Propionic Acid Degradation by Syntrophic Bacteria During Anaerobic Biowaste Digestion by . Monika Felchner-Z wirełło Dissertation, Karlsruher Institut für Technologie (KIT) Fakultät für Fakultät für Bauingenieur-, Geo- und Umweltwissenschaften Tag der mündlichen Prüfung: 08. Februar 2013 Referenten: Prof. Dr. rer. nat. habil. Josef Winter Korreferenten: Prof. Dr.-Ing. E.h. Hermann H. Hahn, Ph.D. Prof. Dr hab. in˙z . Jacek Namie´snik Impressum Karlsruher Institut für Technologie (KIT) KIT Scientific Publishing Straße am Forum 2 D-76131 Karlsruhe KIT Scientific Publishing is a registered trademark of Karlsruhe Institute of Technology. Reprint using the book cover is not allowed. www.ksp.kit.edu This document – excluding the cover – is licensed under the Creative Commons Attribution-Share Alike 3.0 DE License (CC BY-SA 3.0 DE): http://creativecommons.org/licenses/by-sa/3.0/de/ The cover page is licensed under the Creative Commons Attribution-No Derivatives 3.0 DE License (CC BY-ND 3.0 DE): http://creativecommons.org/licenses/by-nd/3.0/de/ Print on Demand 2014 ISSN 1614-5267 ISBN 978-3-7315-0159-6 DOI: 10.5445/KSP/1000037825 Propionic Acid Degradation by Syntrophic Bacteria During Anaerobic Biowaste Digestion Zur Erlangung des akademischen Grades eines DOKTOR-INGENIEURS von der Fakult¨at f¨ur Bauingenieur-, Geo- und Umweltwissenschaften des Karlsruher Instituts f¨ur Technologie (KIT) genehmigte DISSERTATION von Dipl.-Ing. -

'Candidatus Desulfonatronobulbus Propionicus': a First Haloalkaliphilic
Delft University of Technology ‘Candidatus Desulfonatronobulbus propionicus’ a first haloalkaliphilic member of the order Syntrophobacterales from soda lakes Sorokin, D. Y.; Chernyh, N. A. DOI 10.1007/s00792-016-0881-3 Publication date 2016 Document Version Accepted author manuscript Published in Extremophiles: life under extreme conditions Citation (APA) Sorokin, D. Y., & Chernyh, N. A. (2016). ‘Candidatus Desulfonatronobulbus propionicus’: a first haloalkaliphilic member of the order Syntrophobacterales from soda lakes. Extremophiles: life under extreme conditions, 20(6), 895-901. https://doi.org/10.1007/s00792-016-0881-3 Important note To cite this publication, please use the final published version (if applicable). Please check the document version above. Copyright Other than for strictly personal use, it is not permitted to download, forward or distribute the text or part of it, without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license such as Creative Commons. Takedown policy Please contact us and provide details if you believe this document breaches copyrights. We will remove access to the work immediately and investigate your claim. This work is downloaded from Delft University of Technology. For technical reasons the number of authors shown on this cover page is limited to a maximum of 10. Extremophiles DOI 10.1007/s00792-016-0881-3 ORIGINAL PAPER ‘Candidatus Desulfonatronobulbus propionicus’: a first haloalkaliphilic member of the order Syntrophobacterales from soda lakes D. Y. Sorokin1,2 · N. A. Chernyh1 Received: 23 August 2016 / Accepted: 4 October 2016 © Springer Japan 2016 Abstract Propionate can be directly oxidized anaerobi- from its members at the genus level.