1
2 Supplementary Figures
3
SH-PEG-Lactonolactone 55 4 50 45 A 40 35 5 30 25 20 15 10 6 5 %T 554000 3500 3000 2500 2000 1500 1000 500 50 7 45 SH-PEG-Coumarin 40 35 30 B 8 25 20 15 10 9 5 4000 3500 3000 2500 2000 1500 1000 500 10 wavelength (in nm)
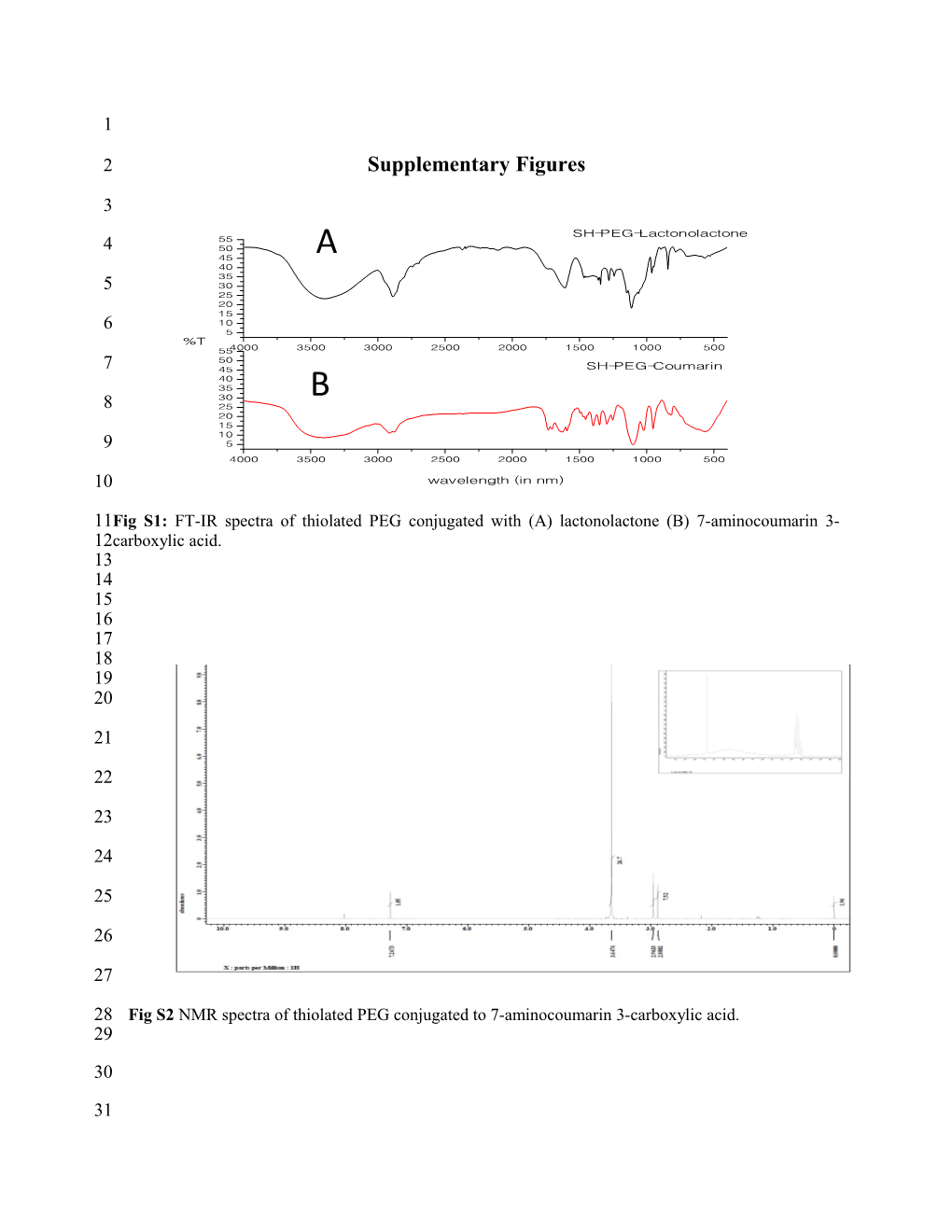
11Fig S1: FT-IR spectra of thiolated PEG conjugated with (A) lactonolactone (B) 7-aminocoumarin 3- 12carboxylic acid. 13 14 15 16 17 18 19 20
21
22
23
24
25
26
27
28 Fig S2 NMR spectra of thiolated PEG conjugated to 7-aminocoumarin 3-carboxylic acid. 29
30
31 32
33 3.0
2.5 34 PEG conjugated Coumarine 2.0 Absorbance intensity 7-aminocoumarin 3-carboxylic acid 35 1.5
1.0 373nm
36 0.5 368nm
0.0 37 300 400 500 600 700 800 900 1000 1100 wavelength (in nm) 38
39
40Fig S3: UV-Vis analysis curve showing hypsochromic shift in λmax value for 7-aminocoumarin 3- 41carboxylic acid and its PEG conjugated derivative 42 43 44 45 46 47 48
49
50
51
52
53
54 FigS4: FT-IR spectra of bare gold nanoparticles 55
56
57
58
59
60
61 1.0 62 522nm Bare Gold Nanoparticles 0.8 63 Bioconjugated Gold nanoparticles 0.6 64 Absorbance 0.4 563nm
65 0.2
66 0.0 67 400 500 600 700 800 68 wavelength (in nm) 69 70 71 72 Fig S5: UV-Vis analysis data showing bathochromic shift in λmax value of gold nanoparticles up on 73 conjugation 74
75
76
77
78 A B 79
80
81
82
83
84 Fig S6: QELS Data (A) and TEM Micrograph (B) of conjugated gold nanoparticles
85
86
87
88
89
90
91 CH2OH CH2OH 92 OH O O OH OH
CH2OH CH2OH O 93 OH OH O O O O OH OH 94 OH OH
95 OH OH
96 Scheme S1: Synthesis of lactonolactone from lactose 97 98
99
100
101
CHO COOH HOOC 102 Aniline/Ethanol 70-80oC + CH2 HOOC 103 CHO O O
Conc H2SO4/ConcHNO3 104 0oC
105
COOH 106
O O 107 O2N
SnCl2/HCl RT 108
COOH 109
O O 110 H2N 111 112 Scheme S2: Synthesis of 7-aminocoumarin 3-carboxylic acid 113 114. 115
116
117
118
119 120
121
122 HO O O OMe 123 n Thioacetate N /RT 124 2
HO O 125 O SAc n
126 Tosylchloride/Et3N 60oC/RT 127 tsO O O SAc n 128 NaN3/RT
129 N N O O SAc n 130 LiAlH4 o 131 0 C Derivative 3. iv H2N O O SH 132 n
133 Scheme S3: - Synthesis of SH-PEG-NH2 from monomethoxy PEG 5000 134
135
136
137
138
139
140
141
142 143
144
145
HO O 146 O OMe n Tosylchloride 147 DMAP+Et3N 60oC
tsO O 148 O OMe Derivative 4. i n DMSO 149 Na2HP4
OHC O O OMe Derivative 4. ii 150 n
151 Thiacetate N2/RT
O 152 OHC O SAc n
NaOMe/MeOH 153 HCl/RT O OHC O SH 154
155
156Scheme S4: Synthesis of SH-PEG-CHO from monomethoxy PEG 2000 157
158
159
160
161
162
163
164
165 166
167
168 CH2OH CH2OH O OH HS O O 169 OH O OH O NH2 O n 170
OH 171 OH CH2OH CH2OH O 172 HO OH O OH HC OH HS O O 173 O N n H
174 OH OH 175
176 Scheme S5:- Coupling of lactonolactone to SH-PEG-NH2 to form Adduct 1 177
178
179
180
181 COOH
182 HS O CHO O O O n H N 183 2 1. Et N 3 184 COOH 2. NaBH CN 3 185 H HS O C O O 186 O NH n 187
188 Scheme S6:- Coupling of Coumarin derivative to SH-PEG-CHO to form Adduct 2 189 190
191 Suppelmentary Methods
1922.2 Synthesis of Targeting Moiety: Lactonolactone
193
194Lactonolactone was synthesised by oxidizing the sugar moiety Lactose17. Briefly, lactose (2g;) was
195dissolved in minimum amount of hot water followed by its addition to an iodine solution in
196methanol(3g/40mL) at 40˚C. The reaction mixture was stirred for 2 hours. Following this, a concentrated
197solution of potassium hydroxide in methanol was added drop-wise to the reaction mixture until the colour
198of iodine disappeared. The solution was then cooled externally in an ice bath which led to the
199precipitation of a crystalline product. The product was filtered, repeatedly washed with cold methanol and
200recrystallized using water/methanol system.
201
202The potassium salt of lactobionic acid thus formed was converted to its free acid form by passing it
203through a column of Acidic amberlite resin. The acidic elute was concentrated and evaporated several
204times with methanol to get the final lactone as a highly viscous colourless oil. The scheme for the
205synthesis of lactonolactone from lactose is shown in Scheme S1. IR spectra: - 3371, 2898, 1736, 1647,
2061421, 1226, 1139, 1077, 1035, 891, 787
207
2082.2.3 Synthesis of Fluorescent moiety: 7-aminocoumarin 3-carboxylic acid
209
210The scheme for the synthesis of fluorescent 7-aminocoumarin 3-carboxylic acid is shown in Scheme S2.
211Synthesis of the fluorescent moiety was performed in three consecutive steps, the first one involving the
212synthesis of 3-carboxycoumarin from Salicyldehyde and Malonic acid as per the procedure discussed by
213Besson et al18, followed by its controlled nitration at cold temperatures to give 7-nitrocoumarin 3- 214carboxylic acid. The later was reduced with SnCl2/HCl mixture to yield the final product as a bright
215yellow solid.
216
217Data for 7-aminocoumarin 3-carboxylic acid: 1H N.M.R: - 8.93(d, COOH), 8.52(d, ArH), 7.65 (d, ArH),
-1 2184.18 (NH2), 3.86 (CH=C) IR spectra (ᶹ/CM ): - 3428, 3369-3400 (d, primary amine), 3069, 2855, 1741
219(C=O for lactone), 1705 (C=O for carboxylic acid), 1610 () N-H), 1096 (C-O), 1001, 947, 845.
220
2212.2.4 Synthesis of SH-PEG-NH2 from monomethoxy PEG 5000
222
223The synthesis of hetero bi-functional PEG derivative from monomethoxy PEG 5000 involves a sequence
224of steps as illustrated in Scheme S3. The synthetic procedures have been referred from a few
225publications19, 20 and were performed with slight modifications.
226
227Derivative 3.i: - Monomethoxy PEG 5000 (1.0Eq) was dissolved in minimum amount of DMF followed
228by the addition of Potassium thioacetate (10.0 Eq) under inert atmosphere. The reaction mixture was
229allowed to stir under nitrogen atmosphere for about 24 hours. The progress of the reaction was monitored
230through TLC (methanol/CH2Cl2 system). After the completion of the reaction, the crude reaction mixture
231was treated with CH2Cl2 and excess thioacetate was washed by adding equal portions of saturated solution
232of NH4Cl and brine. The aqueous and the organic layer were then separated followed by 4-5 times
233extraction of the aqueous layer with CH2Cl2. Finally, the organic layer aliquots were combined, evaporated
234and purified over alumina (methanol/CH2Cl2 system) to get the final crude product as yellow oil with foul
235smell. The yellow oil when triturated with Diethyl ether gave pale yellow solid as the final product. 1H
-1 236N.M.R 2.15 (S, CH3), 3.14 (t, AcS-CH2), 3.60-3.68 (m, CH2-CH2-O-CH2-CH2), IR Spectra (ᶹ/cm ) – 3433
237(br, m), 2888 (br, vs), 1680, 1467, 1343 (asymmetric S=O stretch), 1281, 1250, 1112 (C-O stretch), 963,
238842
239 240Derivative 3.ii - Thioacetate derivative of PEG was tosylated using the procedure as follows: - AcS-PEG-
241OH (1.0eq) was dissolved in minimum amount of toluene followed by addition of base triethylamine
242(3.0eq) and p-tosylchloride (1.5eq). The reaction mixture was stirred for about 5 hrs at a maintained
243temperature of 60˚C after which it was left on stirring for another 10 hrs at ambient temperature. Progress
244of the reaction was monitored through TLC (methanol/CH2Cl2 system). Following the completion of the
245reaction, the solvent was removed from the reaction mixture over Buchi rotary evaporator. The crude
246product was then dissolved in CH2Cl2, and treated with 0.25M aqueous HBr solution and brine which
247served as washing agents for excess triethylamine and tosylchloride, respectively. The aqueous and
248organic layers were separated. The aqueous layer was extracted 4-5 times with CH2Cl2. Finally, the organic
249layer aliquots were combined, evaporated and purified over alumina (Methanol/ CH2Cl2) to get the
250product as pale yellow coloured oil, which upon trituration with ether yielded pale yellow final product. .
1 251 H N.M.R 2.15 (s, S- CH3), 2.42 (s, CH3) 3.12 (t, AcS-CH2), 3.50-3.72 (m, CH2-CH2-O-CH2-CH2), 4.12 (t,
-1 252CH2-Ots), 7.31 (d, ArH), 7.79 (d, ArH) IR Spectra (ᶹ/cm ): - 3400 (absorbed water), 2888, 1735, 1673,
2531466, 1344 (asymmetric S=O stretch), 1281, 1112, 1034(S-O stretch), 980, 842.
254
255Derivative 3.iii - Derivative 3.ii (1.0eq) was dissolved in minimum amount of DMF followed by the
256addition of sodium azide NaN3 (1.25eq) under inert atmosphere at ambient conditions. The reaction
257mixture was allowed to stir at the same conditions for 24hrs. The formation of product was confirmed
258through TLC (CH2Cl2/MeOH). The crude product was then repeatedly precipitated out from the reaction
259mixture by addition of dry ether. Purification by chromatography over alumina (EtOAc/MeOH) yielded
260the final product as pale yellow oil which upon trituration with ether gave pale yellow coloured solid. . 1H
261N.M.R 2.15 (s, S-CH3), 3.14 (t, AcS-CH2), 3.32(t, CH2) 3.53-3.68 (m, CH2-CH2-O-CH2-CH2) IR spectra
262(ᶹ/cm-1): - 2936, 2888, 2098 (azide stretch), 1644, 1466, 1346, 1281, 1110, 1034, 981, 843.
263
264Derivative 3.iv Under complete Argon atmosphere, a solution of LiAlH4 (5.0eq) in dry DMF was stirred
265for about half an hour at -10 to 0˚C (maintained in ice/Sodium chloride bath) in a dry round bottom flask, 266before a solution of Azido-PEG-Thioacetate (1.0eq) in DMF was added drop wise into it. Stirring at the
267maintained conditions was continued for another 4hrs. The progress of the reaction was analysed via
268Ellman’s test for thiol group and ninhydrin test for the amino group. After the completion of the reaction,
269double distilled water was cautiously added. Lithium hydroxide thus precipitated out was filtered over a
270pad of celite and washed repeatedly with ethanol. The filterate was concentrated, dissolved in minimum
271amount of CH2Cl2 and purified over alumina (EtOAc/MeOH system) to yield colourless oil. Trituration
272with dry ether yielded off white coloured Solid. 1.1–1.28 (m, SH), 1.97-2.1 (s, NH 2), 2.23–2.56 (m, CH2),
-1 2733.51–3.76 (m, CH2-CH2-O-CH2-CH2) IR spectra (ᶹ/cm ): - 3369 (strong, primary amine), 2918 (SH
274stretch), 2887, 1598 (N-H bend), 1465, 1346, 1282, 1112, 963
275
276
2772.2.5 Synthesis of SH-PEG-CHO from monomethoxy PEG 2000
278
279A versatile hetero-bifunctional polyethylene glycol (PEG) derivative containing active end-groups thiol
280and aldehyde was efficiently prepared from monomethoxy PEG as per the scheme in scheme S4. Though
281the synthetic procedures were referred from a few publications19, 20, 21, the synthesis was performed with
282considerable modifications.
283
284Derivative 4.i - Purified monomethoxy PEG (1.0eq) was dissolved in minimum amount of toluene
285followed by the addition of Et3N (3.0eq), DMAP (catalytic amount; 0.25eq) and p-tosylchloride (1.5eq).
286The reaction mixture was then heated in an oil bath maintained at 80˚C for 72 hrs. The progress of the
287reaction was analysed through TLC (CH2Cl2/MeOH system). After completion of the reaction, the
288reaction mixture was cooled to room temperature, the solvent was evaporated and the crude oil thus
289obtained was treated with CH2Cl2. Excessive reagents were removed through vigorous washings with
290saturated solution of NaHCO3 & brine and 0.25M aqueous HBr. Finally, the aqueous and organic layers
291were separated. The Aqueous layer was extracted extensively with CH2Cl2. The organic layer aliquots 292were combined, evaporated and purified over alumina (CH2Cl2/MeOH system) to obtain crude final
293product as colourless oil. Trituration with dry ether gave the final product as off white coloured solid. 1H
294N.M.R: - 2.39 (s, ArCH3), 3.32 (s, O-CH3), 3.53-3.66 (m, CH2-CH2-O-CH2-CH2), 4.13 (t, CH2-CH2-Ots),
2957.33 (d, ArH), 7.77(d, ArH) Ir spectra (cm-1): - 3432, 2884, 1735, 1647, 1598, 1466, 1345 (asymmetric
296SO2 stretch), 1281, 1234, (symmetric SO2 stretch), 1112, 949, 842
297Derivative 4.ii - A solution of MeO-PEG-Ots (1.0eq) in 15mL DMSO was treated with Na2HPO4
298(20.0eq) and the mixture was stirred in an oil bath maintained at 100˚C for 20hrs. The progress of the
299reaction was monitored through TLC (CH2Cl2/MeOH system). After the completion of the reaction, the
300cooled reaction mixture was filtered and the filtrate was precipitated out several times with dry ether. The
301precipitate thus obtained was dissolved in minimum amount of water, dialysed and lyophilized to obtain
1 302the final product as an off-white coloured solid. H N.M.R- 3.32 (s, O-CH3), 3.56-3.71 (m, CH2-CH2-O-
-1 303CH2-CH2), 3.81(CH2-CH2-O-CH2-CHO), 4.17 (d CH2-CHO), 9.71(s, CHO) IR spectra (ᶹ/cm ): -3412
304(absorbed water), 2820-2835 (overtone for aldehyde) 2885, 1735 (C=O stretch), 1467, 1254, 1192, 1110,
305953, 847
306
307Derivative 4.iii- CHO-PEG-OMe was subjected to thioacetylation in the next step. To a solution of
308derivative 4.ii (1.0eq) in DMF, 1.5 equivalents of potassium thioacetate were added under inert
309atmosphere. The reaction was allowed to stir continuously at room temperature and nitrogen/argon
310atmosphere for 24 hrs. The progress of the reaction was monitored through TLC (methanol/CH2Cl2
311system). The work up of the reaction mixture was done following the same steps as done for derivative 3.i
1 312which gave the final thioacetated product as a pale yellow solid. H N.M.R- 2.16 (s, CH3), 3. 43(s,
313AcS-CH2), 3.51-3.68 (m, CH2-CH2-O-CH2-CH2), 3.81(CH2-CH2-O-CH2-CHO), 4.17 (d CH2-CHO),
3149.71(s, CHO) IR spectra (ᶹ/cm-1): -3412 (absorbed water), 2820-2835 (overtone for aldehyde) 2885, 1735,
3151687, 1467, 1350, 1254, 1192, 1110, 1034 (S-O strech), 953, 847
316 317
318Derivative 4.iv - To a solution of Derivative 4.iii (1.0eq) in degassed methanol, 5 equivalents of NaOMe
319in MeOH was added. The mixture was allowed to stir overnight at room temperature. Then, the mixture
320was acidified to pH 1–2 using 0.1N HCl. Solvent from the reaction mixture was evaporated over Buchi
321rotary evaporator to give the crude product. Purification by silica gel chromatography (CH 2Cl2/MEOH
322system) gave the bifunctional derivative as colourless oil, which upon trituration with Dry ether gave off-
1 323white coloured solid. H spectra 1.1–1.28 (m, SH), 2.23–2.56 (m, CH2), 3.51–3.76 (m, CH2-CH2-O-CH2-
324CH2), 3.81(CH2-CH2-O-CH2-CHO), 4.17 (d CH2-CHO), 9.71(s, CHO) IR spectra: - 3420 (absorbed
325water), 2918 (SH stretch) 2820-2835 (overtone for aldehyde), 2884, 2110, 1735, 1466, 1351, 1253, 1099,
3261022, 953, 845
327
328
329NOTE: The coupling reactions of the thiolated PEG with Gold nanoparticles, target specific and
330fluorescent moieties were done within 2-3days of their synthesis as the thiol linkages lack stability in
331oxidizing environment and have strong tendency to form disulfides.
332
333