CHAPTER 40 ALTERATIONS OF DIGESTIVE FUNCTION IN CHILDREN
Disorders of the GI tract
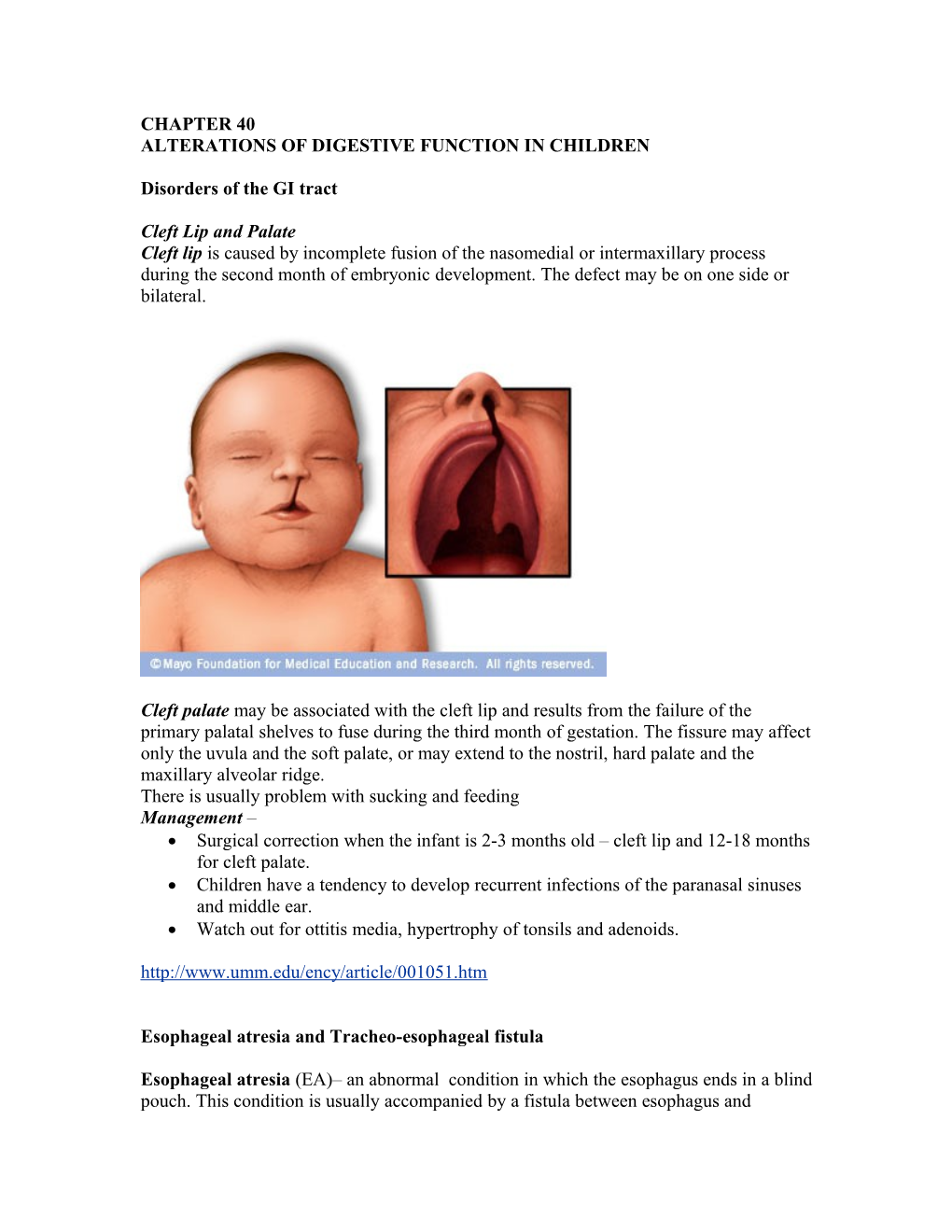
Cleft Lip and Palate Cleft lip is caused by incomplete fusion of the nasomedial or intermaxillary process during the second month of embryonic development. The defect may be on one side or bilateral.
Cleft palate may be associated with the cleft lip and results from the failure of the primary palatal shelves to fuse during the third month of gestation. The fissure may affect only the uvula and the soft palate, or may extend to the nostril, hard palate and the maxillary alveolar ridge. There is usually problem with sucking and feeding Management – Surgical correction when the infant is 2-3 months old – cleft lip and 12-18 months for cleft palate. Children have a tendency to develop recurrent infections of the paranasal sinuses and middle ear. Watch out for ottitis media, hypertrophy of tonsils and adenoids. http://www.umm.edu/ency/article/001051.htm
Esophageal atresia and Tracheo-esophageal fistula
Esophageal atresia (EA)– an abnormal condition in which the esophagus ends in a blind pouch. This condition is usually accompanied by a fistula between esophagus and trachea - tracheo-esophageal fistula (TEF). This is due to defective differentiation as the trachea separates from the esophagus during the 4 – 6 weeks of embryonic development.
EA is suspected in an infant with excessive salivation (drooling) and in a newborn with drooling that is frequently accompanied by choking, coughing and sneezing. When fed, these infants swallow normally but begin to cough and struggle as the fluid returns through the nose and mouth. The infant may become cyanotic and may stop breathing as the overflow of fluid from the blind pouch is aspirated into the trachea resulting in cyanosis as a result of laryngospasm. Over time respiratory distress will develop. If any of the above signs/symptoms are noticed, a catheter is gently passed into the esophagus to check for resistance. If resistance is noted, radiological studies are done to confirm diagnosis.
Treatment of EA and TEF is surgery to repair the defect. The child has to be monitored for pulmonary complication even after surgical correction.
In TEF without EA the baby presents with recurrent symptoms of aspiration, pneumonia, and atelectasis.
PYLORIC STENOSIS It is an obstruction of the pyloric sphincter caused by hypertrophy and hyperplasia of the pyloric sphincter muscle, seen early in infant life (1 – 2 weeks). Associated with Role of transforming growth factor –alpha in stimulating this hypertrophy Increased gastrin secretion by mother in the last trimester of pregnancy Exogenous administration of prostaglandin E Increased incidence in children with Down syndrome Manifestations Vomiting with no apparent reason which turns projectile immediately after feeding Infants are hungry and want to feed again May present with constipation Fluid and electrolyte imbalances – hyperchloremic metabolic alkalosis Chronic malnutrition and weight loss Management Surgical correction – pyloromyotomy
MECONIUM ILEUS
Intestinal obstruction caused by meconium that is abnormally viscous, insoluble, and sticks to the mucosa of the small intestine often associated with cystic fibrosis. It can rarely occur in infants with normal pancreas. There is usually a lack of pancreatic digestive enzymes in fetal life that lead to inspissated meconium that produces obstruction. http://www-medlib.med.utah.edu/WebPath/PEDHTML/PED046.html This meconium is found to contain albumin ( not normal – used to screen for cystic fibrosis). The segment proximal to the obstruction is distended and hypertrophied. The distal segment is collapsed, filled with pale pellets of stool. Normal peristalsis fails to dislodge this impaction and may result in volvulus, atresia, or perforation of the bowels. Manifestations Abdominal distention showing dilated intestinal loops Failure to pass meconium Vomiting Pulmonary symptoms if accompanied with cystic fibrosis Sweat test positive in infants with cystic fibrosis Management Radiological confirmation of the diagnosis Hyperosmolar enema during fluoroscopy to evacuate the meconium Enterotomy HIRSCHSPRUNG DISEASE (HD) - (Congenital Aganglion Megacolon)
Functional obstruction caused by inadequate motility. It is commonly found in Down Syndrome children. It can be life-threatening or a chronic disorder. HD develops before a child is born. Normally, nerve cells grow in the baby's intestine during the fetal life.These nerve cells grow down from the top of the intestine all the way to the anus. With HD, the nerve cells stop growing before they reach the end. The inadequate motility is a result of an aganglionic (without nerve tissue) section of the intestines resulting in megacolon (dilated section of colon). Manifestation Failure to pass meconium in the first week of life Failure to thrive Poor feeding Chronic constipation Vomiting Abdominal distention Management Rectal biopsy is done to diagnose Hirschsprung's Disease Removal of the aganglionic portion of the colon Temporary colostomy to help the child to thrive Reconstructive surgery later with closure of colostomy ANORECTAL MALFORMATIONS Anal stenosis, anal or rectal agenesis, atresia, fistula, and Imperforate anus (Pg 1454 Fig 40 -4)
Intussusception Telescoping of one portion of the intestine into another causing intestinal obstruction; commonly the proximal portion collapses into the distal portion. This causes compression of the mesenteric vessels venous stasis engorgement edema and further compression.
Manifestations Abdominal pain (colicky) Vomiting Currant jelly stools Tender sausage abdominal mass Management Diagnosed on basis of USG and CT abdomen Reduction with a barium enema Surgical reduction
GASTROESOPHAGEAL REFLUX (GER) It is caused by gastric acid flowing from the stomach into the esophagus. GER is often the result of conditions that affect the lower esophageal sphincter, when this muscle relaxes too often or for too long and acid refluxes back into the esophagus, causing vomiting or heartburn.
Manifestations
As feedings are digested, the sphincter opens and allows the stomach contents to go back up the esophagus. Sometimes, the stomach contents go all the way up the esophagus and the baby vomits. Other times, the stomach contents only go part of the way up the esophagus, causing heartburn, breathing problems, or sometimes salient. Sometimes the stomach contents move up the esophagus and spill over into the windpipe, causing asthma, pneumonia, and possibly even SIDS (sudden infant death syndrome). Esophagitis) or ulcers can form in the esophagus due to contact with stomach acid. These can be painful and also may bleed, leading to anemia due to bleeding. Esophageal stricture and Barrett's esophagus (abnormal cells in the esophageal lining) are long term complications from GER.
Management
Mild GR resolves without treatment. Small frequent feeding
Medical management – medications to increase gastric motility, increase lower esophageal sphincter pressure, or decrease gastric acid production
Surgical intervention – gastropexy and fundoplication http://www.healthsystem.virginia.edu/uvahealth/peds_hrnewborn/gerd.cfm
IMPAIRMENT OF DIGESTION, ABSORPTION, AND NUTRITION
CYSTIC FIBROSIS (CF) It is a genetically transmitted condition, that affects the cells that produce mucus, sweat, saliva and digestive juices. Normally, these secretions are thin and slippery, but in cystic fibrosis, a defective gene causes the secretions to become thick and sticky. Instead of acting as a lubricant, the secretions plug up tubes, ducts and passageways, especially in the pancreas and lungs. Respiratory failure is the most dangerous consequence of cystic fibrosis. Also, the secretions block pancreatic enzymes that help digest fats and proteins, and they prevent your body from absorbing key vitamins. The CF triad 1. deficiency of pancreatic enzyme 2. Overproduction of mucous in the respiratory tract – progressive COPD 3. Abnormally elevated sodium and chloride concentrations in the sweat. Manifestations In new born – Meconium ileus may be the first presenting symptom Failure to thrive Steatorrhea Frequent respiratory infections
In children and young adults Salty taste to skin Foul smelling greasy stools Coughing, wheezing, frequent chest infections Rectal prolapse – straining May result in cirrhosis as a result of blockage of the cystic ducts Management The main goal is to prevent infections, reduce the amount and thickness of secretions in the lungs, improve airflow, and maintain adequate calories and nutrition (Pancreatic enzyme replacement, High protein high calorie diet).
This link is very helpful. Please go through this interactive tutorial http://www.nlm.nih.gov/medlineplus/tutorials/cysticfibrosis/htm/index.htm
GLUTEN SENSITIVE ENTEROPATHY - Celiac Disease (CD) It is an autoimmune intestinal disorder, found in individuals who are genetically susceptible. Damage to the mucosal surface of the small intestine is caused by an immunologically toxic reaction to the ingestion of gluten and interferes with the absorption of nutrients. When individuals with CD ingest gluten, the villi in the small intestine that absorb nutrients from food, are damaged. This is due to an immunological reaction to gluten. Damaged villi do not effectively absorb basic nutrients -- proteins, carbohydrates, fats, vitamins, minerals, and, in some cases, water and bile salts. If CD is left untreated, damage to the small bowel can be chronic and life threatening, causing an increased risk of associated disorders -- both nutritional and immune related.
Cause: Current research indicates that CD is strongly associated with a group of genes on Chromosome 6. These genes (HLA class II antigens) are involved in the regulation of the body's immune response to the gluten protein fractions
Manifestations: http://digestive.niddk.nih.gov/ddiseases/pubs/celiac/index.htm#2
Management: http://www.mayoclinic.com/health/celiacdisease/DS00319/DSECTION=8
PROTEIN ENERGY MALNUTRITION- PEM Kwashiokar and Marasmus – two most common types of malnutrition in children; disorders collectively known as PEM . The wet form is called kwashiorkor (severe protein deficiency), an African word meaning "first child-second child." It refers to the observation that the first child develops PEM when the second child is born and replaces the first child at the breast. The weaned child is fed a thin gruel of poor nutritional quality (compared with mother's milk) and fails to thrive. The lack of sufficient plasma proteins, particularly albumin, causes systemic pressure changes that result in generalized edema, increase in the volume of total body water and extra cellular fluid, and loss of potassium. The liver swells with stored fat as there is no synthesis of hepatic proteins. This further leads to further mal absorption and malnutrition. The protein deficiency is usually more marked than the energy deficiency. Marasmus – severe deficiency of all nutrients, leading to reduced metabolic processes. In marasmus, energy intake is insufficient for the body's requirements, and the body draws on its own stores. Liver glycogen is exhausted within a few hours, and skeletal muscle protein is then used via gluconeogenesis to maintain adequate plasma glucose. At the same time, triglycerides in fat depots are broken down into free fatty acids, which provide some energy for most tissues, but not for the nervous system. When near starvation is prolonged, fatty acids are incompletely oxidized to ketone bodies, which can be used by the brain and other organs for energy. Thus, in the severe energy deficiency of marasmus, adaptation is facilitated by high cortisol and growth hormone levels and depression of insulin and thyroid hormone secretion. Because amino acids are mobilized from muscle to provide the liver with substrate for protein synthesis, plasma protein levels decrease less in marasmus than in kwashiorkor. Marasmus differs from kwashiorkor in several important aspects: Marasmus Kwashiorkor
1. The onset is earlier, usually Onset is later, after the breast-feeding in the first year of life is stopped.
2. Growth failure is more Not very Pronounced. pronounced.
3. There is no edema Edema is present.
4. Blood protein concentration Blood protein concentration is reduced is reduced less markedly. very much.
5. Skin changes are seen less Red boils and patches are classic frequently. symptoms.
6. Liver is not infiltrated with Fatty liver is seen. fat
7. Recovery is much longer. Recovery period is short. http://images.google.com/imgres?imgurl=http://www.who.int/child-adolescent- health/publications/referral_care/chap7/marasmus.gif&imgrefurl=http://www.who.int/chi ld-adolescent- health/publications/referral_care/chap7/chap71.htm&h=400&w=220&sz=6&tbnid=7Rtf Niy86id3tM:&tbnh=124&tbnw=68&prev=/images%3Fq %3Dmarasmus&start=2&sa=X&oi=images&ct=image&cd=2 http://www.merck.com/mrkshared/mmanual/section1/chapter2/2c.jsp NECROTIZING ENTEROCOLITIS
Necrotizing enterocolitis is an acquired disease, primarily in premature infants or sick newborns, in which intestinal tissue dies. The cause for this disorder is unknown, but it is thought that a decreased blood flow to the bowel keeps the bowel from producing the normal protective mucus. Bacteria in the intestine may also be an added cause causing accumulation of gas in the mucosa and sub-mucosa leading to ischemia, inflammation and necrosis of intestinal segments.
Radiological aspect of NEC with Pneumatosis Intestinalis and portal air
Manifestation: Abdominal distention Vomiting and feeding intolerance Blood in the stool (visible or microscopic) Lethargy Temperature instability Diarrhea Management With infants suspected of NEC, stop feedings and gas in the bowel is relieved IV fluids to replace feeds Antibiotic therapy Continuous monitoring (abdominal x-rays, blood tests, and blood gases) In case of intestinal peritonitis, perforation surgical correction (colostomy, ileostomy)
DISORDERS OF THE LIVER
Biliary Atresia
Biliary atresia is an obstruction of the bile ducts caused by their failure to develop normally in the fetus. This is a congenital condition (present at birth). Biliary atresia is caused by the abnormal development of the bile ducts inside or outside the liver. The purpose of the biliary system is to remove waste products from the liver, and to carry bile salts necessary for fat digestion to the small intestine.In babies with biliary atresia, bile flow from the liver to the gallbladder is blocked. This can lead to liver damage and cirrhosis of liver, which, if not treated, will eventually be fatal. http://www.nlm.nih.gov/medlineplus/ency/article/001145.htm
Symptoms of biliary atresia.
Babies with biliary atresia usually appear healthy when they are born. Symptoms of the disease typically appear within the first two weeks to two months of life. Jaundice -- a yellow coloring of the skin and eyes due to a very high level of bilirubin (bile pigment) in the bloodstream.
Jaundice caused by an immature liver is common in newborns. It usually goes away within the first week to 10 days of life. A baby with biliary atresia usually appears normal at birth, but develops jaundice at two or three weeks after birth.
Dark urine -- caused by the build-up of bilirubin (a breakdown product from hemoglobin) in the blood. The bilirubin is then filtered by the kidney and removed in the urine.
Acholic stools (clay-colored stools) -- because no bile or bilirubin coloring is being emptied into the intestine. Also, the abdomen may become swollen from a firm, enlarged liver.
Weight loss and irritability -- develop when the level of jaundice increases.
Diagnosis and management: http://www.orpha.net/nestasso/OFAVB/__PP__4.html WILSON DISEASE
Wilson's Disease is an autosomal recessive disease. The responsible gene is located at a precisely known site on chromosome 13, ATP7B. Some cases of Wilson's Disease occur due to spontaneous mutations in the gene. Most are transmitted from generation to generation.
Wilson's Disease is a genetic disorder that is fatal unless detected and treated before serious illness from copper poisoning develops. The body to retain copper. As the intestines absorb copper from food, the copper builds up in the liver and injures liver tissue. Eventually, the damage causes the liver to release the copper directly into the bloodstream, which carries the copper throughout the body. The copper buildup leads to damage in the kidneys, brain, and eyes. If not treated, Wilson's disease can cause severe brain damage, liver failure, and death. Manifestation A triad of neuromuscular abnormalities, intentional tremors, dysarthria, and dystonia: 1. Kayser-Fleischer rings (accumulation of copper in the limbus of the cornea, causing a greenish yellow ring)
2. Cirrhosis associated with elevated serum copper 3. low ceruloplasmin levels Initially the child may just present with malaise, abdominal pain and jaundice. It may later progress to edema, GI bleed or a hemolytic crisis. If untreated cirrhosis develops.
If untreated children will die of neural, hepatic, renal, or hematologic complications http://digestive.niddk.nih.gov/ddiseases/pubs/wilson/index.htm http://www.medstudents.com.br/metdis/metdis2.htm
Congenital anomalies of GI http://www.centrus.com.br/DiplomaFMF/SeriesFMF/18-23-weeks/chapter- 07/gifmf.html