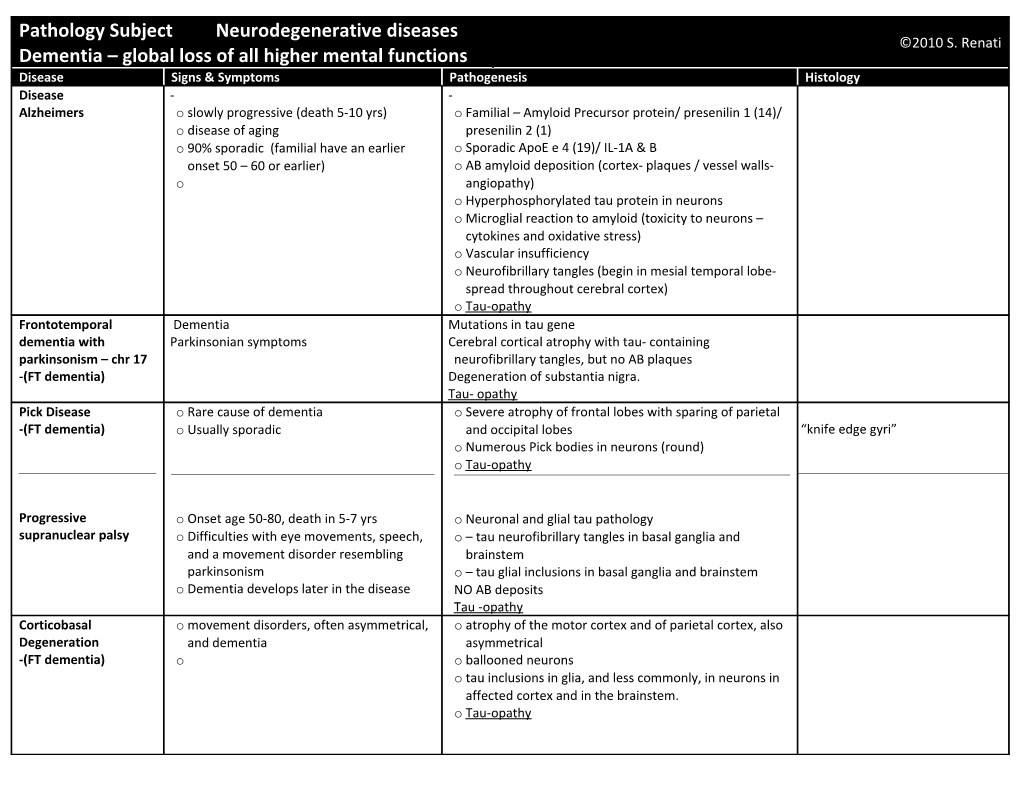
Pathology Subject Neurodegenerative diseases ©2010 S. Renati Dementia – global loss of all higher mental functions Disease Signs & Symptoms Pathogenesis Histology Disease - - Alzheimers o slowly progressive (death 5-10 yrs) o Familial – Amyloid Precursor protein/ presenilin 1 (14)/ o disease of aging presenilin 2 (1) o 90% sporadic (familial have an earlier o Sporadic ApoE e 4 (19)/ IL-1A & B onset 50 – 60 or earlier) o AB amyloid deposition (cortex- plaques / vessel walls- o angiopathy) o Hyperphosphorylated tau protein in neurons o Microglial reaction to amyloid (toxicity to neurons – cytokines and oxidative stress) o Vascular insufficiency o Neurofibrillary tangles (begin in mesial temporal lobe- spread throughout cerebral cortex) o Tau-opathy Frontotemporal Dementia Mutations in tau gene dementia with Parkinsonian symptoms Cerebral cortical atrophy with tau- containing parkinsonism – chr 17 neurofibrillary tangles, but no AB plaques -(FT dementia) Degeneration of substantia nigra. Tau- opathy Pick Disease o Rare cause of dementia o Severe atrophy of frontal lobes with sparing of parietal -(FT dementia) o Usually sporadic and occipital lobes “knife edge gyri” o Numerous Pick bodies in neurons (round) o Tau-opathy
Progressive o Onset age 50-80, death in 5-7 yrs o Neuronal and glial tau pathology supranuclear palsy o Difficulties with eye movements, speech, o – tau neurofibrillary tangles in basal ganglia and and a movement disorder resembling brainstem parkinsonism o – tau glial inclusions in basal ganglia and brainstem o Dementia develops later in the disease NO AB deposits Tau -opathy Corticobasal o movement disorders, often asymmetrical, o atrophy of the motor cortex and of parietal cortex, also Degeneration and dementia asymmetrical -(FT dementia) o o ballooned neurons o tau inclusions in glia, and less commonly, in neurons in affected cortex and in the brainstem. o Tau-opathy Motor neuron disease o may or may not have clinical motor o small ubiquitin positive neuronal inclusions with no Ubiquitin + inclusion in fascia inclusion dementia neuron disease other pathology dentata of hippocampus -(FT dementia) o may or may not be genetic o (picture in notes) o In familial cases some family members may have dementia, while others may have motor neuron disease
Dementia lacking o o Superficial cortical distinctive histology microvasculization is common to -(FT dementia) many FTDs
Vascular dementia o Clinically distinguished by a “stepwise” o Generally caused by numerous small strokes progression. Although not always reliable o Synergistic with alzheimers: less AD path required for dementia if vascular disease present
Parkinson’s Disease o Slowed movements and rigidity o Lewy bodies in substantia nigra Lewy bodies in substantia nigra o Tremor o Death of substantia nigra dopaminergic neurons o Preservation of higher cortical functions in o Loss of normal balance in basal ganglia circuits most cases o A-syncleinopathy o Some pts. Show psychiatric changes and progressive dementia: Dementia with Lewy bodies o Most cases sporadic o Familial -> genes-> a-synuclein / Parkin
Dementia with Lewy o Dementing disease similar to Alzheimer’s, o Always Lewy bodies in SN, but there is not always Always Lewy bodies in SN bodies but often with hallucinations and parkinsonism fluctuations in symptoms o A-synucleinopathy o Overlap with Alz. Disease: most (60-70%) DLB cases also show ALZ. pathology Multiple system o Three normally separate diseases now o The common pathology is a-synuclein inclusions in atrophy united under the term MSA b/c of their affected areas. The inclusions are primarily in common a-synuclein pathology Oligodendrocytes. o – striatonigral degeneration o A- synucleinopathy o –olivepontocerebellar atrophy o shy-drager syndrome
Huntington’s disease o Age of onset varies (30-50) o CAG trinucleotide repeats in Huntington gene (chr 4) o Autosomal Dominant o – abnormal Huntington protein with glutamine repeats Atrophy of caudate nucleus and o Large, involuntary, “dance-like” at one end putamen movements(chorea) loss of regulation o – impairs mitochondrial function and axonal transport of cortical motor neurons o – worsening in successive generations o Later, dementia o Degeneration of straite nuclei (caudate / putamen) o o – loss of neurons, inhib GABA neurons are particularly affected o –leads to disruption of normal inhib/excit balance in basal ganglia circuits o Later, cortical atrophy with loss of neurons
Spinocerebellar ataxia o A group of diseases all genetic o degeneration of spinal cord and cerebellar neurons and o Most are dominant, some recessive tracts o o Friedrich ataxia begins in childhood, death w/in 5 yrs. Associated with heart disease and diabetes. GAA repeats in frataxin gene o Ataxia-telangiectasis begins in early childhood, death by age 20. telangiectasias of conjunctiva, skin and CNS. Abnormal response to DNA damage: continued replication w/o repair or apoptosis (immunodef. w/ recurrent infections and various cancers.
Leucodystrophies o Long tract signs – ataxia/ pyramidal signs. o Diffuse degeneration of CNS whire matter due to o Clinical presentation is dominated bny malformed myelin motor signs rather than cognitive decline: o Krabbe (globoid cell) leucodystrophy spasticity, hypotonia, ataxia o --- deficiency of galactocerebroside B-galactosidase Krabbe cell : perivascular o Most have onset in early childhood: an o --- inability to degrade galactocerebroside macrophages (globoid cells) are exception is adrenoleucodystrophy o --- alternate catabolism generates galactosphingosine, distended with cerebroside) wh/ is toxic to oligodendrocytes o --- macrophages collect undigested cerebroside, and form multinucleated giant cells around the blood vessel o Metachromatic leucodystrophy o ---arylsulfatase A deficiency o --- defective degeneration of sulfatides o – there is a (rare) adult form that can present with psych. Symp or with progressive dementia o --- Accumulated sulfatides stain red-brown w/ cresyl violet o Adrenoleucodystrophy o --- peroxismal defect, x-linked o --- inability to degrade fatty acids with more than 22 carbons (very long chain fatty acids) Clefts in Macrophages in ALD o --- there is also atrophy of the adrenal glands with accum. Of VLCFA o ---most common in 5-9yro children o adult form with progressive paraplegic of legs (adrenomyeloneuropathy) Mitochondrial diseases o involve tissues with high aerobic demands o muscle, heart, retina, and brain o primarily diseases of young adults
MERRF MELAS KEARNS SAYRE
------ Leigh disease Fatal disease of early childhood Caused by various mutations affecting cytochrome c Peculiar degeneration of brain tissue adj. to CSF pathways with sparing of neurons Pathology resembles Wernicke-Korsakoff although the clinical setting is entirely different. Vit Def: Thiamine (B1) o wernike encephalopathy: an acute o May cause peripheral neuropathy (beri beri) psychotic/opthalmoplegic syndrome o May cause degeneration of mamillary bodies and of Acute W-K disease: note o korsakoff syndrome: chronic memory brain tissue adj. to CSF pathways (WK synd) that hemorrahagic mamillary bodies disturbances with confabulation resembles Leigh disease o W-K syndrome is most commonly seen in association with cachexia or poor nutrition: alcoholism/ GI disease/cancer Vit. Deficiencies: B12 o Numbness and tingling of legs, proceeding o Degeneration of both ascending and descending tracts to spastic weakness and paraplegia of spinal cord: subacute combined degeneration of the Myelin loss in lateral and posterior o Folate def can cause a similar syndrome spinal cord columns
Metabolic disorders o o hypoglycemia similar to hypoxic injury o hyperglycemia dehydration affects brain function/ overly rapid rehydration can cause cerebral edema o Hepatic encephalopathy hyperammonenia leads to confusion progressing o coma/ Histologically there are altered astrocytes (alz. Type 2 glia) in the cerebral cortex Toxic disorders o o Carbon Monoxide similar to hypoxic injury/ bilateral necrosis of globus pallidus can occur/ delayed demyelination can occur o Methanol retinal degeneration/ bilateral necrosis of putamen o Ethanol massive, acute ethanol intoxication can cause cerebral swelling and death/ it is unclear alone has any chronic effects on CNS/ associated with nutritional deficiencies can cause cerebellar vermal atrophy or Wk syndrome/ fetal exposure results in severe deficits o Radiation radionecrosis of brain is due to endothelial injury with fibrinoid degeneration of vessel and thrombosis/ this can follow radiotherapy for malignant brain tumor o Combined methotrexate and radiation injury white matter necrosis/ may occur months after exposure