Advances in Therapeutic Targeting of Cancer Stem Cells Within the Tumor Microenvironment: an Updated Review
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Stem Cells and Cancer
Stem Cells and Cancer Cancer Education Project Stem Cells and Cancer Overview: This series of activities is designed to introduce students to the theory that some cancers arise from cancer stem cells. This theory provides a possible explanation for why cancers reoccur after cancer treatment. It also provides insights that may lead to new types of chemotherapy drugs. • Part 1: Stem Cells and Cancer PowerPoint (40 minutes) Students view a PowerPoint presentation that introduces stem cell biology and shows ways that cancer stem cell research might lead to more effective cancer therapy treatments. Students answer questions as they view the PowerPoint. Then they create a cartoon strip to illustrate their understanding of cancer stem cells. • Part 2: The Bad Seed: Rare stem cells appear to drive cancers (20 minutes) Students read a brief article that introduces stem cell biology and explains how cancer stem cell research might lead to more effective cancer therapy treatments. Students answer questions based on this article. This activity may be done in class or for homework. • Part 3: Plant Derivative Attacks the Roots of Leukemia (20 minutes) Students read a brief article on the development of a potential chemotherapy agent that specifically targets cancer stem cells. Students answer questions based on this article. This activity may be done in class or for homework. • Part 4: Clinical Trials: Parthocet (40 minutes) Students answer questions about the design of a large-scale, randomized, double-blind clinical trial to determine if Parthocet (a fictitious chemotherapy drug) is safe and effective. Life Sciences Learning Center – Cancer Education Project 1 Copyright © 2007, University of Rochester May be copied for classroom use Stem Cells and Cancer Teacher Instructions - Part 1 Stem Cells and Cancer PowerPoint Presentation Students view a PowerPoint presentation that introduces stem cell biology and shows ways that cancer stem cell research might lead to more effective cancer therapy treatments. -

A Folding Switch Regulates Interleukin 27 Biogenesis and Secretion of Its Α-Subunit As a Cytokine
A folding switch regulates interleukin 27 biogenesis and secretion of its α-subunit as a cytokine Stephanie I. Müllera,b, Antonie Friedlc, Isabel Aschenbrennera,b, Julia Esser-von Bierenc, Martin Zachariasd, Odile Devergnee,1, and Matthias J. Feigea,b,1 aCenter for Integrated Protein Science, Department of Chemistry, Technical University of Munich, 85748 Garching, Germany; bInstitute for Advanced Study, Technical University of Munich, 85748 Garching, Germany; cCenter of Allergy and Environment (ZAUM), Technical University of Munich and Helmholtz Center Munich, 80802 Munich, Germany; dCenter for Integrated Protein Science, Physics Department, Technical University of Munich, 85748 Garching, Germany; and eSorbonne Université, INSERM, CNRS, Centre d’Immunologie et des Maladies Infectieuses, 75 013 Paris, France Edited by John J. O’Shea, NIH, Bethesda, MD, and accepted by Editorial Board Member Tadatsugu Taniguchi December 10, 2018 (received for review October 3, 2018) A common design principle of heteromeric signaling proteins is the by the secretion and biological activity of some isolated α-and use of shared subunits. This allows encoding of complex messages β-subunits (10, 11). A prominent example is IL-27α/p28. Murine IL- while maintaining evolutionary flexibility. How cells regulate and 27α, also designated as IL-30, is secreted in isolation (12) and per- control assembly of such composite signaling proteins remains an forms immunoregulatory roles (13–16). In contrast, no autonomous important open question. An example of particular complexity and secretion of human IL-27α has been reported yet. The molecular biological relevance is the interleukin 12 (IL-12) family. Four func- basis for this difference has remained unclear, but it is likely to have tionally distinct αβ heterodimers are assembled from only five sub- a profound impact on immune system function, since in mice IL-27 units to regulate immune cell function and development. -

Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected]
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School July 2017 Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the Pathology Commons Scholar Commons Citation Mohamed, Mai, "Role of Amylase in Ovarian Cancer" (2017). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/6907 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Role of Amylase in Ovarian Cancer by Mai Mohamed A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Pathology and Cell Biology Morsani College of Medicine University of South Florida Major Professor: Patricia Kruk, Ph.D. Paula C. Bickford, Ph.D. Meera Nanjundan, Ph.D. Marzenna Wiranowska, Ph.D. Lauri Wright, Ph.D. Date of Approval: June 29, 2017 Keywords: ovarian cancer, amylase, computational analyses, glycocalyx, cellular invasion Copyright © 2017, Mai Mohamed Dedication This dissertation is dedicated to my parents, Ahmed and Fatma, who have always stressed the importance of education, and, throughout my education, have been my strongest source of encouragement and support. They always believed in me and I am eternally grateful to them. I would also like to thank my brothers, Mohamed and Hussien, and my sister, Mariam. I would also like to thank my husband, Ahmed. -

Ncounter® Mouse Autoimmune Profiling Panel - Gene and Probe Details
nCounter® Mouse AutoImmune Profiling Panel - Gene and Probe Details Official Symbol Accession Alias / Previous Symbol Official Full Name Other targets or Isoform Information AW208573,CD143,expressed sequence AW208573,MGD-MRK- Ace NM_009598.1 1032,MGI:2144508 angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 2610036I19Rik,2610510L13Rik,Acinus,apoptotic chromatin condensation inducer in the nucleus,C79325,expressed sequence C79325,MGI:1913562,MGI:1919776,MGI:2145862,mKIAA0670,RIKEN cDNA Acin1 NM_001085472.2 2610036I19 gene,RIKEN cDNA 2610510L13 gene apoptotic chromatin condensation inducer 1 Acp5 NM_001102405.1 MGD-MRK-1052,TRACP,TRAP acid phosphatase 5, tartrate resistant 2310066K23Rik,AA960180,AI851923,Arp1b,expressed sequence AA960180,expressed sequence AI851923,MGI:2138136,MGI:2138359,RIKEN Actr1b NM_146107.2 cDNA 2310066K23 gene ARP1 actin-related protein 1B, centractin beta Adam17 NM_001277266.1 CD156b,Tace,tumor necrosis factor-alpha converting enzyme a disintegrin and metallopeptidase domain 17 ADAR1,Adar1p110,Adar1p150,AV242451,expressed sequence Adar NM_001038587.3 AV242451,MGI:2139942,mZaADAR adenosine deaminase, RNA-specific Adora2a NM_009630.2 A2AAR,A2aR,A2a, Rs,AA2AR,MGD-MRK-16163 adenosine A2a receptor Ager NM_007425.2 RAGE advanced glycosylation end product-specific receptor AI265500,angiotensin precursor,Aogen,expressed sequence AI265500,MGD- Agt NM_007428.3 MRK-1192,MGI:2142488,Serpina8 angiotensinogen (serpin peptidase inhibitor, clade A, member 8) Ah,Ahh,Ahre,aromatic hydrocarbon responsiveness,aryl hydrocarbon -

Cancer Stem Cells and Nucleolin As Drivers of Carcinogenesis
pharmaceuticals Review Cancer Stem Cells and Nucleolin as Drivers of Carcinogenesis Laura Sofia Carvalho 1,Nélio Gonçalves 1 , Nuno André Fonseca 1,2 and João Nuno Moreira 1,3,* 1 CNC—Center for Neurosciences and Cell Biology, Center for Innovative Biomedicine and Biotechnology (CIBB), Faculty of Medicine (Polo 1), University of Coimbra, Rua Larga, 3004-504 Coimbra, Portugal; laurasofi[email protected] (L.S.C.); [email protected] (N.G.); [email protected] (N.A.F.) 2 TREAT U, SA—Parque Industrial de Taveiro, Lote 44, 3045-508 Coimbra, Portugal 3 UC—University of Coimbra, CIBB, Faculty of Pharmacy (FFUC), Pólo das Ciências da Saúde, Azinhaga de Santa Comba, 3000-548 Coimbra, Portugal * Correspondence: [email protected]; Tel.: +351-239-820-190 Abstract: Cancer, one of the most mortal diseases worldwide, is characterized by the gain of specific features and cellular heterogeneity. Clonal evolution is an established theory to explain heterogeneity, but the discovery of cancer stem cells expanded the concept to include the hierarchical growth and plasticity of cancer cells. The activation of epithelial-to-mesenchymal transition and its molecular players are widely correlated with the presence of cancer stem cells in tumors. Moreover, the acquisition of certain oncological features may be partially attributed to alterations in the levels, location or function of nucleolin, a multifunctional protein involved in several cellular processes. This review aims at integrating the established hallmarks of cancer with the plasticity of cancer cells as an emerging hallmark; responsible for tumor heterogeneity; therapy resistance and relapse. The discussion will contextualize the involvement of nucleolin in the establishment of cancer hallmarks and its application as a marker protein for targeted anticancer therapies Keywords: tumor heterogeneity; drug resistance; cancer stem cells; nucleolin; targeted therapies; epithelial-to-mesenchymal transition Citation: Carvalho, L.S.; Gonçalves, N.; Fonseca, N.A.; Moreira, J.N. -

Interleukin-30/Il27p28 Shapes Prostate Cancer Stem-Like Cell Behavior and Is Critical for Tumor Onset and Metastasization Carlo Sorrentino1,2,3, Stefania L
Published OnlineFirst February 27, 2018; DOI: 10.1158/0008-5472.CAN-17-3117 Cancer Tumor Biology and Immunology Research Interleukin-30/IL27p28 Shapes Prostate Cancer Stem-like Cell Behavior and Is Critical for Tumor Onset and Metastasization Carlo Sorrentino1,2,3, Stefania L. Ciummo1,2,3, Giuseppe Cipollone4,5, Sara Caputo6, Matteo Bellone6, and Emma Di Carlo1,2,3 Abstract Prostate cancer stem-like cells (PCSLC) are believed to be signaling. IL30 overproduction by PCSLCs promoted tumor responsible for prostate cancer onset and metastasis. Autocrine onset and development associated with increased proliferation, and microenvironmental signals dictate PCSLC behavior and vascularization, and myeloid cell recruitment. Furthermore, it patient outcome. In prostate cancer patients, IL30/IL27p28 has promoted PCSLC dissemination to lymph nodes and bone been linked with tumor progression, but the mechanisms marrow by upregulating the CXCR5/CXCL13 axis, and drove underlying this link remain mostly elusive. Here, we asked metastasis to lungs through the CXCR4/CXCL12 axis. These whether IL30 may favor prostate cancer progression by condi- mechanisms were drastically hindered by IL30 knockdown tioning PCSLCs and assessed the value of blocking IL30 to or knockout in PCSLCs. Collectively, these results mark IL30 suppress tumor growth. IL30 was produced by PCSLCs in as a key driver of PCSLC behavior. Targeting IL30 signaling may human and murine prostatic intraepithelial neoplasia and be a potential therapeutic strategy against prostate cancer pro- displayed significant autocrine and paracrine effects. PCSLC- gression and recurrence. derived IL30 supported PCSLC viability, self-renewal and Significance: IL30 plays an important role in regulating pro- tumorigenicity, expression of inflammatory mediators and state cancer stem-like cell behavior and metastatic potential, growth factors, tumor immune evasion, and regulated chemo- therefore targeting this cytokine could hamper prostate cancer kine and chemokine receptor genes, primarily via STAT1/STAT3 progression or recurrence. -

Cancer from the Perspective of Stem Cells and Misappropriated Tissue Regeneration Mechanisms
Leukemia (2018) 32:2519–2526 https://doi.org/10.1038/s41375-018-0294-7 REVIEW ARTICLE Corrected: Correction Stem cell biology Cancer from the perspective of stem cells and misappropriated tissue regeneration mechanisms 1,2 1 1 1,2 1 Mariusz Z. Ratajczak ● Kamila Bujko ● Aaron Mack ● Magda Kucia ● Janina Ratajczak Received: 8 September 2018 / Accepted: 17 September 2018 / Published online: 30 October 2018 © The Author(s) 2018. This article is published with open access Abstract Tumorigenesis can be considered as pathologically misappropriated tissue regeneration. In this review we will address some unresolved issues that support this concept. First, we will address the issue of the identity of cancer-initiating cells and the presence of cancer stem cells in growing tumors. We will also ask are there rare and distinct populations of cancer stem cells in established tumor cell lines, or are all of the cells cancer stem cells? Second, the most important clinical problem with cancer is its metastasis, and here a challenging question arises: by employing radio-chemotherapy for tumor treatment, do we unintentionally create a prometastatic microenvironment in collateral organs? Specifically, many factors upregulated in response to radio-chemotherapy-induced injury may attract highly migratory cancer cells that survived initial treatment. 1234567890();,: 1234567890();,: Third, what is the contribution of normal circulating stem cells to the growing malignancy? Do circulating normal stem cells recognize a tumor as a hypoxia-damaged tissue that needs -

Wsx-1/Il-27 As a Target for Anti-Inflammatory Responses
(19) TZZ ZZ_T (11) EP 2 046 809 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07K 1/00 (2006.01) C07K 14/54 (2006.01) 07.12.2016 Bulletin 2016/49 C07K 14/715 (2006.01) C07K 16/24 (2006.01) (21) Application number: 07836143.3 (86) International application number: PCT/US2007/016329 (22) Date of filing: 18.07.2007 (87) International publication number: WO 2008/011081 (24.01.2008 Gazette 2008/04) (54) WSX-1/IL-27 AS A TARGET FOR ANTI-INFLAMMATORY RESPONSES WSX-1/IL-27 ALS TARGET FÜR ENTZÜNDUNGSHEMMENDE REAKTIONEN WSX-1/IL-27 UTILISÉ COMME CIBLE POUR SUSCITER DES RÉACTIONS ANTI-INFLAMMATOIRES (84) Designated Contracting States: • VILLARINO ALEJANDRO V ET AL: "IL-27 limits AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IL-2 production during Th1 differentiation." HU IE IS IT LI LT LU LV MC MT NL PL PT RO SE JOURNALOF IMMUNOLOGY (BALTIMORE, MD. : SI SK TR 1950) 1 JAN 2006, vol. 176, no. 1, 1 January 2006 Designated Extension States: (2006-01-01), pages 237-247, XP002570061 ISSN: AL BA RS 0022-1767 • KASTELEIN ROBERT A ET AL: "Discovery and (30) Priority: 19.07.2006 US 832213 P biologyof IL-23 andIL-27: relatedbut functionally 11.08.2006 US 837450 P distinct regulators of inflammation." ANNUAL REVIEW OF IMMUNOLOGY 2007, vol. 25, 2007, (43) Date of publication of application: pages 221-242, XP002570062 ISSN: 0732-0582 15.04.2009 Bulletin 2009/16 • SCHELLER J ET AL: "No inhibition of IL-27 signaling by soluble gp130" BIOCHEMICAL AND (73) Proprietor: The Trustees of The University of BIOPHYSICAL RESEARCH COMMUNICATIONS, Pennsylvania ACADEMIC PRESS INC. -

GP130 Cytokines in Breast Cancer and Bone
cancers Review GP130 Cytokines in Breast Cancer and Bone Tolu Omokehinde 1,2 and Rachelle W. Johnson 1,2,3,* 1 Program in Cancer Biology, Vanderbilt University, Nashville, TN 37232, USA; [email protected] 2 Vanderbilt Center for Bone Biology, Department of Medicine, Division of Clinical Pharmacology, Vanderbilt University Medical Center, Nashville, TN 37232, USA 3 Department of Medicine, Division of Clinical Pharmacology, Vanderbilt University Medical Center, Nashville, TN 37232, USA * Correspondence: [email protected]; Tel.: +1-615-875-8965 Received: 14 December 2019; Accepted: 29 January 2020; Published: 31 January 2020 Abstract: Breast cancer cells have a high predilection for skeletal homing, where they may either induce osteolytic bone destruction or enter a latency period in which they remain quiescent. Breast cancer cells produce and encounter autocrine and paracrine cytokine signals in the bone microenvironment, which can influence their behavior in multiple ways. For example, these signals can promote the survival and dormancy of bone-disseminated cancer cells or stimulate proliferation. The interleukin-6 (IL-6) cytokine family, defined by its use of the glycoprotein 130 (gp130) co-receptor, includes interleukin-11 (IL-11), leukemia inhibitory factor (LIF), oncostatin M (OSM), ciliary neurotrophic factor (CNTF), and cardiotrophin-1 (CT-1), among others. These cytokines are known to have overlapping pleiotropic functions in different cell types and are important for cross-talk between bone-resident cells. IL-6 cytokines have also been implicated in the progression and metastasis of breast, prostate, lung, and cervical cancer, highlighting the importance of these cytokines in the tumor–bone microenvironment. This review will describe the role of these cytokines in skeletal remodeling and cancer progression both within and outside of the bone microenvironment. -
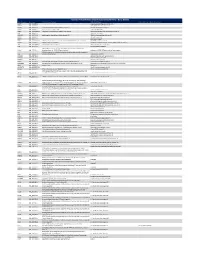
Ncounter® Autoimmune Discovery Consortium Panel
nCounter® Autoimmune Discovery Consortium Panel - Gene Details Official Symbol Accession Alias / Previous Symbol Official Full Name Other targets or Isoform Information AAMP NM_001087.3 angio associated migratory cell protein ABHD6 NM_020676.5 abhydrolase domain containing 6 ACKR2 NM_001296.3 CMKBR9,CCBP2;chemokine binding protein 2 atypical chemokine receptor 2 ACOXL NM_018308.1 acyl-Coenzyme A oxidase-like acyl-CoA oxidase like ACSL6 NM_001009185.1 FACL6;fatty-acid-Coenzyme A ligase, long-chain 6 acyl-CoA synthetase long chain family member 6 ADA NM_000022.2 adenosine deaminase ADAM30 NM_021794.2 a disintegrin and metalloproteinase domain 30 ADAM metallopeptidase domain 30 ADCY3 NM_004036.3 adenylate cyclase 3 ADCY7 NM_001114.4 adenylate cyclase 7 AFF3 NM_001025108.1 LAF4;lymphoid nuclear protein related to AF4,AF4/FMR2 family, member 3 AF4/FMR2 family member 3 AGAP2 NM_014770.3 CENTG1;centaurin, gamma 1 ArfGAP with GTPase domain, ankyrin repeat and PH domain 2 AHI1 NM_001134830.1 Abelson helper integration site Abelson helper integration site 1 AHR NM_001621.3 aryl hydrocarbon receptor AHSA2;AHA1, activator of heat shock 90kDa protein ATPase homolog 2 AHSA2 NM_152392.1 (yeast),activator of HSP90 ATPase homolog 2 activator of HSP90 ATPase homolog 2, pseudogene APECED;autoimmune regulator (autoimmune polyendocrinopathy candidiasis AIRE NM_000383.2 ectodermal dystrophy) autoimmune regulator AMIGO3 NM_198722.2 adhesion molecule with Ig like domain 3 ANKRD55 NM_024669.2 ankyrin repeat domain 55 ANTXR2 NM_058172.5 anthrax toxin receptor -

Cancer Stem Cells: a New Approach to Tumor Development
REVIEW ARTICLE KOBAYASHI NCC ET AL. Cancer stem cells: a new approach to tumor development NATÁLIA CRISTINA CIUFA KOBAYASHI1*, SAMUEL MARCOS RIBEIRO DE NORONHA2 1Full Teaching Degree in Biological Sciences – Graduate degree in Molecular Biology, United Metropolitan Colleges (FMU), São Paulo, SP, Brazil 2PhD – Department of Allergy and Immunology, University of São Paulo (USP), São Paulo, SP, Brazil SUMMARY Many theories have been proposed to explain the origins of cancer. Currently, evidences show that not every tumor cell is capable of initiating a tumor. Only a small part of the cancer cells, called cancer stem cells (CSCs), can generate a tumor identical to the original one, when removed from human tumors and transplanted into immunosuppressed mice. The name given to these cells co- mes from the resemblance to normal stem cells, except for the fact that their abi- lity to divide is infinite. These cells are also affected by their microenvironment. Many of the signaling pathways, such as Wnt, Notch and Hedgehog, are altered in this tumoral subpopulation, which also contributes to abnormal prolifera- tion. Researchers have found several markers for CSCs; however, much remains to be studied, or perhaps a universal marker does not even exist, since they vary Study conducted at United among tumor types and even from patient to patient. It was also found that Metropolitan Colleges (FMU) cancer stem cells are resistant to radiotherapy and chemotherapy. This may ex- Article received: 4/24/2014 plain the re-emergence of the disease, since they are not completely eliminated Accepted for publication: 4/24/2014 and minimal amounts of CSCs can repopulate a tumor. -

Cancer Cell CD44 Mediates Macrophage/Monocyte-Driven Regulation of Head and Neck Cancer Stem Cells
Author Manuscript Published OnlineFirst on August 14, 2020; DOI: 10.1158/0008-5472.CAN-20-1079 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Article Cancer cell CD44 mediates macrophage/monocyte-driven regulation of head and neck cancer stem cells. Authors Karina E. Gomez1, FangLong Wu2,3, Stephen B. Keysar1, J. Jason Morton1, Bettina Miller1, Tugs-Saikhan Chimed1, Phuong N. Le1, Cera Nieto1, Farshad N. Chowdhury1, Anit Tyagi1, Traci R. Lyons1,4, Christian D. Young2, Hongmei Zhou3, Hilary L. Somerset2, Xiao-Jing Wang2,4,5, and Antonio Jimeno1,4. Affiliation 1 Division of Medical Oncology, Department of Medicine, University of Colorado Anschutz Medical Campus (CU AMC), CO, 80045. 2 Department of Pathology, CU AMC, CO, 80045. 3 State Key Laboratory of Oral Diseases, Department of Oral Medicine, West China Hospital of Stomatology, Sichuan University, Chengdu, China. 4 Gates Center for Regenerative Medicine, CU AMC, CO, 80045. 5 Veterans Affairs Medical Center, VA Eastern Colorado Health Care System, Aurora, CO, USA. Corresponding Author Antonio Jimeno M.D., Ph.D., Professor of Medicine/Oncology, and Otolaryngology. University of Colorado Cancer Center, and Gates Center for Regenerative Medicine. University of Colorado Anschutz Medical Campus. 12801 East 17th Avenue, Room L18-8101B, Aurora, CO 80045, USA. [email protected] 1 Downloaded from cancerres.aacrjournals.org on September 28, 2021. © 2020 American Association for Cancer Research. Author Manuscript Published OnlineFirst on August 14, 2020; DOI: 10.1158/0008-5472.CAN-20-1079 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.