Assembly Rules of the Microbiome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Microbiology of Endodontic Infections
Scient Open Journal of Dental and Oral Health Access Exploring the World of Science ISSN: 2369-4475 Short Communication Microbiology of Endodontic Infections This article was published in the following Scient Open Access Journal: Journal of Dental and Oral Health Received August 30, 2016; Accepted September 05, 2016; Published September 12, 2016 Harpreet Singh* Abstract Department of Conservative Dentistry & Endodontics, Gian Sagar Dental College, Patiala, Punjab, India Root canal system acts as a ‘privileged sanctuary’ for the growth and survival of endodontic microbiota. This is attributed to the special environment which the microbes get inside the root canals and several other associated factors. Although a variety of microbes have been isolated from the root canal system, bacteria are the most common ones found to be associated with Endodontic infections. This article gives an in-depth view of the microbiology involved in endodontic infections during its different stages. Keywords: Bacteria, Endodontic, Infection, Microbiology Introduction Microorganisms play an unequivocal role in infecting root canal system. Endodontic infections are different from the other oral infections in the fact that they occur in an environment which is closed to begin with since the root canal system is an enclosed one, surrounded by hard tissues all around [1,2]. Most of the diseases of dental pulp and periradicular tissues are associated with microorganisms [3]. Endodontic infections occur and progress when the root canal system gets exposed to the oral environment by one reason or the other and simultaneously when there is fall in the body’s immune when the ingress is from a carious lesion or a traumatic injury to the coronal tooth structure.response [4].However, To begin the with, issue the if notmicrobes taken arecare confined of, ultimately to the leadsintra-radicular to the egress region of pathogensIn total, and bacteria their by-productsdetected from from the the oral apical cavity foramen fall into to 13 the separate periradicular phyla, tissues. -

The Oral Microbiome of Healthy Japanese People at the Age of 90
applied sciences Article The Oral Microbiome of Healthy Japanese People at the Age of 90 Yoshiaki Nomura 1,* , Erika Kakuta 2, Noboru Kaneko 3, Kaname Nohno 3, Akihiro Yoshihara 4 and Nobuhiro Hanada 1 1 Department of Translational Research, Tsurumi University School of Dental Medicine, Kanagawa 230-8501, Japan; [email protected] 2 Department of Oral bacteriology, Tsurumi University School of Dental Medicine, Kanagawa 230-8501, Japan; [email protected] 3 Division of Preventive Dentistry, Faculty of Dentistry and Graduate School of Medical and Dental Science, Niigata University, Niigata 951-8514, Japan; [email protected] (N.K.); [email protected] (K.N.) 4 Division of Oral Science for Health Promotion, Faculty of Dentistry and Graduate School of Medical and Dental Science, Niigata University, Niigata 951-8514, Japan; [email protected] * Correspondence: [email protected]; Tel.: +81-45-580-8462 Received: 19 August 2020; Accepted: 15 September 2020; Published: 16 September 2020 Abstract: For a healthy oral cavity, maintaining a healthy microbiome is essential. However, data on healthy microbiomes are not sufficient. To determine the nature of the core microbiome, the oral-microbiome structure was analyzed using pyrosequencing data. Saliva samples were obtained from healthy 90-year-old participants who attended the 20-year follow-up Niigata cohort study. A total of 85 people participated in the health checkups. The study population consisted of 40 male and 45 female participants. Stimulated saliva samples were obtained by chewing paraffin wax for 5 min. The V3–V4 hypervariable regions of the 16S ribosomal RNA (rRNA) gene were amplified by PCR. -

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -

Actinomyces Naeslundii: an Uncommon Cause of Endocarditis
Hindawi Publishing Corporation Case Reports in Infectious Diseases Volume 2015, Article ID 602462, 4 pages http://dx.doi.org/10.1155/2015/602462 Case Report Actinomyces naeslundii: An Uncommon Cause of Endocarditis Christopher D. Cortes,1 Carl Urban,1,2 and Glenn Turett1 1 The Dr. James J. Rahal, Jr. Division of Infectious Diseases, NewYork-Presbyterian Queens, Flushing, NY 11355, USA 2Department of Medicine, Weill Cornell Medical College, New York, NY 10065, USA Correspondence should be addressed to Carl Urban; [email protected] and Glenn Turett; [email protected] Received 21 September 2015; Accepted 16 November 2015 Academic Editor: Sandeep Dogra Copyright © 2015 Christopher D. Cortes et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Actinomyces rarely causes endocarditis with 25 well-described cases reported in the literature in the past 75 years. We present a case of prosthetic valve endocarditis (PVE) caused by Actinomyces naeslundii.Toourknowledge,thisisthefirstreportintheliterature of endocarditis due to this organism and the second report of PVE caused by Actinomyces. 1. Introduction holosystolic decrescendo murmur. Abnormal laboratory tests included a WBC count of 16.5 K/L (reference range: 4.8– Actinomyces naeslundii is a Gram positive anaerobic bacillary 10.8 K/L) with 88.5% neutrophils and C-reactive protein commensal of the human oral cavity. Septicemia with this (CRP) 7.02 mg/L (reference range: 0.03–0.49 mg/dL). Only organism is uncommon and poses an increased risk of one set of blood cultures was drawn prior to empirically subacute and chronic granulomatous inflammation, which starting vancomycin and ceftriaxone (the patient had a can affect all organ systems via hematogenous spread [1]. -

A Metagenomic Β-Glucuronidase Uncovers a Core Adaptive Function of the Human Intestinal Microbiome
A metagenomic β-glucuronidase uncovers a core adaptive function of the human intestinal microbiome Karine Gloux, Olivier Berteau, Hanane El oumami, Fabienne Béguet, Marion Leclerc, and Joël Doré1 Institut National de la Recherche Agronomique, Unité Mixte de Recherche 1319 Micalis, F-78352 Jouy en Josas, France Edited by Todd R. Klaenhammer, North Carolina State University, Raleigh, NC, and approved June 1, 2010 (received for review February 4, 2010) In the human gastrointestinal tract, bacterial β-D-glucuronidases (BG; XIVa), major reservoirs of BG-positive bacteria (11), are dys- E.C. 3.2.1.31) are involved both in xenobiotic metabolism and in some biosis markers in Crohn’s disease (12–15) and in intestinal carci- of the beneficial effects of dietary compounds. Despite their biolog- nogenesis (16). Nevertheless, the relative contribution of bacterial ical significance, investigations are hampered by the fact that only strains to the global intestinal BG activity remains unclear, and a few BGs have so far been studied. A functional metagenomic the molecular bases are essentially unknown. approach was therefore performed on intestinal metagenomic In the human digestive tract, bacterial BGs are known to be libraries using chromogenic glucuronides as probes. Using this strat- distributed among the Enterobacteriaceae family in some Firmi- egy, 19 positive metagenomic clones were identified but only one cutes genera (Lactobacillus, Streptococcus, Clostridium, Rumino- exhibited strong β-D-glucuronidase activity when subcloned into an coccus, Roseburia, and Faecalibacterium) and in a specific species expression vector. The cloned gene encoded a β-D-glucuronidase of Actinobacteria (Bifidobacterium dentium) (11, 17, 18). On (called H11G11-BG) that had distant amino acid sequence homolo- the basis of their sequence, BGs are highly homologous to gies and an additional C terminus domain compared with known β-galactosidases and only a few of them have been investigated β fi -D-glucuronidases. -
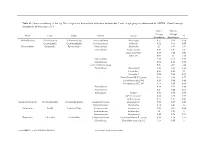
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Download Publication
Caesar Kleberg A Publication of the Caesar Kleberg Wildlife ResearchTracks Institute CAESAR KLEBERG WILDLIFE RESEARCH INSTITUTE TEXAS A&M UNIVERSITY - KINGSVILLE 1 Caesar Kleberg Volume 5 | Issue 2 | Fall 2020 In This Issue 3 From the Director Tracks 4 Restoration: Just What Do You Mean By That? 8 The Unique Nature of Colonial-nesting Waterbirds 12 Texas Horned Lizard 8 Detectives Learn More About CKWRI The Caesar Kleberg Wildlife Research Institute at 16 Donor Spotlight: Texas A&M University-Kingsville is a Master’s and Mike Reynolds Ph.D. Program and is the leading wildlife research organization in Texas and one of the finest in the nation. Established in 1981 by a grant from the Caesar Kleberg 20 Fawn Survival and Foundation for Wildlife Conservation, its mission is Recruitment in South Texas to provide science-based information for enhancing the conservation and management of Texas wildlife. 23 Alumni Spotlight: J. Dale James Visit our Website www.ckwri.tamuk.edu Caesar Kleberg Wildlife Research Institute Texas A&M University-Kingsville 700 University Blvd., MSC 218 Kingsville, Texas 78363 4 (361) 593-3922 Follow us! Facebook: @CKWRI Twitter: @CKWRI Instagram: ckwri_official Cover Photo by iStock 2 Magazine Design and Layout by Gina Cavazos Caesar Kleberg From the Director The overarching mission of the Caesar Kleberg Wildlife Research Institute is to promote wildlife conservation. We do this primarily by conducting research and producing knowledge to benefit wildlife managers and land stewards. We also promote wildlife conservation by training the next generation of wildlife biologists Tracks through our graduate education programs. However, producing knowledge and trained professionals is not enough. -

The Isolation of Novel Lachnospiraceae Strains and the Evaluation of Their Potential Roles in Colonization Resistance Against Clostridium Difficile
The isolation of novel Lachnospiraceae strains and the evaluation of their potential roles in colonization resistance against Clostridium difficile Diane Yuan Wang Honors Thesis in Biology Department of Ecology and Evolutionary Biology College of Literature, Science, & the Arts University of Michigan, Ann Arbor April 1st, 2014 Sponsor: Vincent B. Young, M.D., Ph.D. Associate Professor of Internal Medicine Associate Professor of Microbiology and Immunology Medical School Co-Sponsor: Aaron A. King, Ph.D. Associate Professor of Ecology & Evolutionary Associate Professor of Mathematics College of Literature, Science, & the Arts Reader: Blaise R. Boles, Ph.D. Assistant Professor of Molecular, Cellular and Developmental Biology College of Literature, Science, & the Arts 1 Table of Contents Abstract 3 Introduction 4 Clostridium difficile 4 Colonization Resistance 5 Lachnospiraceae 6 Objectives 7 Materials & Methods 9 Sample Collection 9 Bacterial Isolation and Selective Growth Conditions 9 Design of Lachnospiraceae 16S rRNA-encoding gene primers 9 DNA extraction and 16S ribosomal rRNA-encoding gene sequencing 10 Phylogenetic analyses 11 Direct inhibition 11 Bile salt hydrolase (BSH) detection 12 PCR assay for bile acid 7α-dehydroxylase detection 12 Tables & Figures Table 1 13 Table 2 15 Table 3 21 Table 4 25 Figure 1 16 Figure 2 19 Figure 3 20 Figure 4 24 Figure 5 26 Results 14 Isolation of novel Lachnospiraceae strains 14 Direct inhibition 17 Bile acid physiology 22 Discussion 27 Acknowledgments 33 References 34 2 Abstract Background: Antibiotic disruption of the gastrointestinal tract’s indigenous microbiota can lead to one of the most common nosocomial infections, Clostridium difficile, which has an annual cost exceeding $4.8 billion dollars. -

Streptococcus Adjacens and Streptococcus Defectivus to Abiotrophia Gen
INTERNATIONAL JOURNALOF SYSTEMATIC BACTERIOLOGY, OCt. 1995, p. 798-803 Vol. 45, No. 4 0020-7713/95/$04.00 -t-0 Copyright 0 1995, International Union of Microbiological Societies Transfer of Streptococcus adjacens and Streptococcus defectivus to Abiotrophia gen. nov. as Abiotrophia adiacens comb. nov. and Abiotrophia defectiva comb. nov., Respectively YQSHIAKI KAWAMURA,” XIAO-GANG HOU, FERDOUSI SULTANA, SHUJUN LIU, HTROYUKI YAMAMOTO, AND TAKAYUKI EZAKI Department of Microbiology, Gifu University School of Medicine, 40 Tsukasa-machi, Gifu 500, Japan We performed this study to determine the 16s rRNA sequences of the type strains of Streptococcus adjacens and Streptococcus defectivus and to calculate the phylogenetic distances between these two nutritionally variant streptococci (NVS) and other members of the genus Streptococcus. S. adjacens and S. defectivus belonged to one cluster, but this cluster was not closely related to other streptococcal species. A comparative analysis of the sequences of these organisms and other low-G+C-content gram-positive bacteria revealed that the two NVS species formed a distinct cluster and were only remotely related to the Aerococcus and Carnobacterium clusters. The highest level of homology (93.7%) was found between S. adjacens and Carnobacterium divergens. Carnobac- terium species have meso-diaminopimelic acid in their cell walls, but S. adjacens and S. defectivus have L-lysine as the diamino acid at position 3 in their peptidoglycan tetrapeptides. On the basis of our findings and the results of previous phenotypic studies, we propose that the NVS species should be placed in a new genus, the genus Abiotrophia, as Abiotrophia adiacens comb. nov. and Abiotrophia defectiva comb. -

Fatal Subdural Empyema Caused by Streptococcus Constellatus and Actinomyces Viscosus in a Childdcase Report
Journal of Microbiology, Immunology and Infection (2011) 44, 394e396 available at www.sciencedirect.com journal homepage: www.e-jmii.com CASE REPORT Fatal subdural empyema caused by Streptococcus constellatus and Actinomyces viscosus in a childdCase report Asma Bouziri a,*, Ammar Khaldi a, Hane`ne Smaoui b, Khaled Menif a, Nejla Ben Jaballah a a Pediatric Intensive Care Unit, Children’s Hospital of Tunis, Place Bab Saadoun, Tunis, Tunisia b Department of Microbiology, Children’s Hospital of Tunis, Tunis, Tunisia Received 4 August 2009; received in revised form 21 January 2010; accepted 21 March 2010 KEYWORDS Group milleri streptococci that colonize the mouth and the upper airways are generally Actinomyces viscosus; considered to be commensal. In combination with anaerobics, they are rarely responsible Empyema; for brain abscesses in patients with certain predisposing factors. Mortality in such cases Streptococcus is high and complications are frequent. We present a case of fatal subdural empyema constellatus; caused by Streptococcus constellatus and Actinomyces viscosus in a previously healthy Subdural 7-year-old girl. Copyright ª 2011, Taiwan Society of Microbiology. Published by Elsevier Taiwan LLC. All rights reserved. Introduction intestinal, and urogenital tract.1 After translocation into otherwise sterile sites, they may cause purulent infections Group milleri streptococci (GMS) comprise a heterogeneous and abscess formation mostly among seriously compro- group of streptococci, including the species intermedius, mised patients. Actinomyces -

Gut Dysbiosis and Neurobehavioral Alterations in Rats Exposed to Silver Nanoparticles Received: 10 January 2017 Angela B
www.nature.com/scientificreports OPEN Gut Dysbiosis and Neurobehavioral Alterations in Rats Exposed to Silver Nanoparticles Received: 10 January 2017 Angela B. Javurek1, Dhananjay Suresh 2, William G. Spollen3,4, Marcia L. Hart5, Sarah Accepted: 19 April 2017 A. Hansen6, Mark R. Ellersieck7, Nathan J. Bivens 8, Scott A. Givan 3,4,9, Anandhi Published: xx xx xxxx Upendran10,11, Raghuraman Kannan11,12 & Cheryl S. Rosenfeld 4,13,14,15 Due to their antimicrobial properties, silver nanoparticles (AgNPs) are being used in non-edible and edible consumer products. It is not clear though if exposure to these chemicals can exert toxic effects on the host and gut microbiome. Conflicting studies have been reported on whether AgNPs result in gut dysbiosis and other changes within the host. We sought to examine whether exposure of Sprague- Dawley male rats for two weeks to different shapes of AgNPs, cube (AgNC) and sphere (AgNS) affects gut microbiota, select behaviors, and induces histopathological changes in the gastrointestinal system and brain. In the elevated plus maze (EPM), AgNS-exposed rats showed greater number of entries into closed arms and center compared to controls and those exposed to AgNC. AgNS and AgNC treated groups had select reductions in gut microbiota relative to controls. Clostridium spp., Bacteroides uniformis, Christensenellaceae, and Coprococcus eutactus were decreased in AgNC exposed group, whereas, Oscillospira spp., Dehalobacterium spp., Peptococcaeceae, Corynebacterium spp., Aggregatibacter pneumotropica were reduced in AgNS exposed group. Bacterial reductions correlated with select behavioral changes measured in the EPM. No significant histopathological changes were evident in the gastrointestinal system or brain. Findings suggest short-term exposure to AgNS or AgNC can lead to behavioral and gut microbiome changes.