WO 2009/143603 Al
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of Retinopathy in Type 1 Diabetes
Genetic Analysis of Retinopathy in Type 1 Diabetes by Sayed Mohsen Hosseini A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by S. Mohsen Hosseini 2014 Genetic Analysis of Retinopathy in Type 1 Diabetes Sayed Mohsen Hosseini Doctor of Philosophy Institute of Medical Science University of Toronto 2014 Abstract Diabetic retinopathy (DR) is a leading cause of blindness worldwide. Several lines of evidence suggest a genetic contribution to the risk of DR; however, no genetic variant has shown convincing association with DR in genome-wide association studies (GWAS). To identify common polymorphisms associated with DR, meta-GWAS were performed in three type 1 diabetes cohorts of White subjects: Diabetes Complications and Control Trial (DCCT, n=1304), Wisconsin Epidemiologic Study of Diabetic Retinopathy (WESDR, n=603) and Renin-Angiotensin System Study (RASS, n=239). Severe (SDR) and mild (MDR) retinopathy outcomes were defined based on repeated fundus photographs in each study graded for retinopathy severity on the Early Treatment Diabetic Retinopathy Study (ETDRS) scale. Multivariable models accounted for glycemia (measured by A1C), diabetes duration and other relevant covariates in the association analyses of additive genotypes with SDR and MDR. Fixed-effects meta- analysis was used to combine the results of GWAS performed separately in WESDR, ii RASS and subgroups of DCCT, defined by cohort and treatment group. Top association signals were prioritized for replication, based on previous supporting knowledge from the literature, followed by replication in three independent white T1D studies: Genesis-GeneDiab (n=502), Steno (n=936) and FinnDiane (n=2194). -

Bayesian Hierarchical Modeling of High-Throughput Genomic Data with Applications to Cancer Bioinformatics and Stem Cell Differentiation
BAYESIAN HIERARCHICAL MODELING OF HIGH-THROUGHPUT GENOMIC DATA WITH APPLICATIONS TO CANCER BIOINFORMATICS AND STEM CELL DIFFERENTIATION by Keegan D. Korthauer A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Statistics) at the UNIVERSITY OF WISCONSIN–MADISON 2015 Date of final oral examination: 05/04/15 The dissertation is approved by the following members of the Final Oral Committee: Christina Kendziorski, Professor, Biostatistics and Medical Informatics Michael A. Newton, Professor, Statistics Sunduz Kele¸s,Professor, Biostatistics and Medical Informatics Sijian Wang, Associate Professor, Biostatistics and Medical Informatics Michael N. Gould, Professor, Oncology © Copyright by Keegan D. Korthauer 2015 All Rights Reserved i in memory of my grandparents Ma and Pa FL Grandma and John ii ACKNOWLEDGMENTS First and foremost, I am deeply grateful to my thesis advisor Christina Kendziorski for her invaluable advice, enthusiastic support, and unending patience throughout my time at UW-Madison. She has provided sound wisdom on everything from methodological principles to the intricacies of academic research. I especially appreciate that she has always encouraged me to eke out my own path and I attribute a great deal of credit to her for the successes I have achieved thus far. I also owe special thanks to my committee member Professor Michael Newton, who guided me through one of my first collaborative research experiences and has continued to provide key advice on my thesis research. I am also indebted to the other members of my thesis committee, Professor Sunduz Kele¸s,Professor Sijian Wang, and Professor Michael Gould, whose valuable comments, questions, and suggestions have greatly improved this dissertation. -

Role of Mitochondrial Ribosomal Protein S18-2 in Cancerogenesis and in Regulation of Stemness and Differentiation
From THE DEPARTMENT OF MICROBIOLOGY TUMOR AND CELL BIOLOGY (MTC) Karolinska Institutet, Stockholm, Sweden ROLE OF MITOCHONDRIAL RIBOSOMAL PROTEIN S18-2 IN CANCEROGENESIS AND IN REGULATION OF STEMNESS AND DIFFERENTIATION Muhammad Mushtaq Stockholm 2017 All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by E-Print AB 2017 © Muhammad Mushtaq, 2017 ISBN 978-91-7676-697-2 Role of Mitochondrial Ribosomal Protein S18-2 in Cancerogenesis and in Regulation of Stemness and Differentiation THESIS FOR DOCTORAL DEGREE (Ph.D.) By Muhammad Mushtaq Principal Supervisor: Faculty Opponent: Associate Professor Elena Kashuba Professor Pramod Kumar Srivastava Karolinska Institutet University of Connecticut Department of Microbiology Tumor and Cell Center for Immunotherapy of Cancer and Biology (MTC) Infectious Diseases Co-supervisor(s): Examination Board: Professor Sonia Lain Professor Ola Söderberg Karolinska Institutet Uppsala University Department of Microbiology Tumor and Cell Department of Immunology, Genetics and Biology (MTC) Pathology (IGP) Professor George Klein Professor Boris Zhivotovsky Karolinska Institutet Karolinska Institutet Department of Microbiology Tumor and Cell Institute of Environmental Medicine (IMM) Biology (MTC) Professor Lars-Gunnar Larsson Karolinska Institutet Department of Microbiology Tumor and Cell Biology (MTC) Dedicated to my parents ABSTRACT Mitochondria carry their own ribosomes (mitoribosomes) for the translation of mRNA encoded by mitochondrial DNA. The architecture of mitoribosomes is mainly composed of mitochondrial ribosomal proteins (MRPs), which are encoded by nuclear genomic DNA. Emerging experimental evidences reveal that several MRPs are multifunctional and they exhibit important extra-mitochondrial functions, such as involvement in apoptosis, protein biosynthesis and signal transduction. Dysregulations of the MRPs are associated with severe pathological conditions, including cancer. -

New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology
Cancer Research and Treatment 2003;35(4):304-313 New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology Yong-Wan Kim, Ph.D.1, Min-Je Suh, M.S.1, Jin-Sik Bae, M.S.1, Su Mi Bae, M.S.1, Joo Hee Yoon, M.D.2, Soo Young Hur, M.D.2, Jae Hoon Kim, M.D.2, Duck Young Ro, M.D.2, Joon Mo Lee, M.D.2, Sung Eun Namkoong, M.D.2, Chong Kook Kim, Ph.D.3 and Woong Shick Ahn, M.D.2 1Catholic Research Institutes of Medical Science, 2Department of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul; 3College of Pharmacy, Seoul National University, Seoul, Korea Purpose: This study utilized both mRNA differential significant genes of unknown function affected by the display and the Gene Ontology (GO) analysis to char- HPV-16-derived pathway. The GO analysis suggested that acterize the multiple interactions of a number of genes the cervical cancer cells underwent repression of the with gene expression profiles involved in the HPV-16- cancer-specific cell adhesive properties. Also, genes induced cervical carcinogenesis. belonging to DNA metabolism, such as DNA repair and Materials and Methods: mRNA differential displays, replication, were strongly down-regulated, whereas sig- with HPV-16 positive cervical cancer cell line (SiHa), and nificant increases were shown in the protein degradation normal human keratinocyte cell line (HaCaT) as a con- and synthesis. trol, were used. Each human gene has several biological Conclusion: The GO analysis can overcome the com- functions in the Gene Ontology; therefore, several func- plexity of the gene expression profile of the HPV-16- tions of each gene were chosen to establish a powerful associated pathway, identify several cancer-specific cel- cervical carcinogenesis pathway. -

A Novel Computational Algorithm for Predicting Immune Cell Types Using Single-Cell RNA Sequencing Data
A novel computational algorithm for predicting immune cell types using single-cell RNA sequencing data By Shuo Jia A hesis submitted to the Faculty of Graduate Studies of The University of Manitoba n partial fulfillment of the requirements of the degree of MASTER OF SCIENCE Department of Biochemistry and Medical Genetics University of Manitoba Winnipeg, Manitoba, Canada Copyright © 2020 by Shuo Jia Abstract Background: Cells from our immune system detect and kill pathogens to protect our body against many diseases. However, current methods for determining cell types have some major limitations, such as being time-consuming and with low throughput rate, etc. These problems stack up and hinder the deep exploration of cellular heterogeneity. Immune cells that are associated with cancer tissues play a critical role in revealing the stages of tumor development. Identifying the immune composition within tumor microenvironments in a timely manner will be helpful to improve clinical prognosis and therapeutic management for cancer. Single-cell RNA sequencing (scRNA-seq), an RNA sequencing (RNA-seq) technique that focuses on a single cell level, has provided us with the ability to conduct cell type classification. Although unsupervised clustering approaches are the major methods for analyzing scRNA-seq datasets, their results vary among studies with different input parameters and sizes. However, in supervised machine learning methods, information loss and low prediction accuracy are the key limitations. Methods and Results: Genes in the human genome align to chromosomes in a particular order. Hence, we hypothesize incorporating this information into our model will potentially improve the cell type classification performance. In order to utilize gene positional information, we introduce chromosome-based neural network, namely ChrNet, a novel chromosome-specific re-trainable supervised learning method based on a one-dimensional 1 convolutional neural network (1D-CNN). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

New Resources for Transcription Analysis and Genome Fugu
Downloaded from genome.cshlp.org on July 6, 2011 - Published by Cold Spring Harbor Laboratory Press Fugu ESTs: New Resources for Transcription Analysis and Genome Annotation Melody S. Clark, Yvonne J.K. Edwards, Dan Peterson, et al. Genome Res. 2003 13: 2747-2753 Access the most recent version at doi:10.1101/gr.1691503 References This article cites 51 articles, 26 of which can be accessed free at: http://genome.cshlp.org/content/13/12/2747.full.html#ref-list-1 Article cited in: http://genome.cshlp.org/content/13/12/2747.full.html#related-urls Email alerting Receive free email alerts when new articles cite this article - sign up in the box at the service top right corner of the article or click here To subscribe to Genome Research go to: http://genome.cshlp.org/subscriptions Cold Spring Harbor Laboratory Press Downloaded from genome.cshlp.org on July 6, 2011 - Published by Cold Spring Harbor Laboratory Press Resource Fugu ESTs: New Resources for Transcription Analysis and Genome Annotation Melody S. Clark,1,7,8 Yvonne J.K. Edwards,1 Dan Peterson,2 Sandra W. Clifton,2 Amanda J. Thompson,1 Masahide Sasaki,3 Yutaka Suzuki,3 Kiyoshi Kikuchi,5,6 Shugo Watabe,5 Koichi Kawakami,4 Sumio Sugano,3 Greg Elgar,1 and Stephen L. Johnson2 1MRC Rosalind Franklin Centre for Genomics Research, (formerly known as the MRC UK HGMP Resource Centre), Genome Campus, Hinxton, Cambridge, CB10 1SB, UK; 2Department of Genetics, Washington University Medical School, St Louis, Missouri 63110, USA; 3The Institute of Medical Science, The University of Tokyo, Shirokanedai, -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Gene Expression Effects of Lithium and Valproic Acid in a Serotonergic Cell Line
bioRxiv preprint doi: https://doi.org/10.1101/227652; this version posted December 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Gene expression effects of lithium and valproic acid in a serotonergic cell line. Diana Balasubramanian1, John F. Pearson2, Martin A. Kennedy1 1Gene Structure and Function Laboratory and Carney Centre for Pharmacogenomics, Department of Pathology, University of Otago, Christchurch, New Zealand2 2Biostatistics and Computational Biology unit, University of Otago, Christchurch. Correspondence to: Prof. M. A. Kennedy Department of Pathology University of Otago, Christchurch Christchurch, New Zealand Email: [email protected] Keywords: RNA-Seq, valproic acid, gene expression, mood stabilizer, pharmacogenomics bioRxiv preprint doi: https://doi.org/10.1101/227652; this version posted December 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Valproic acid (VPA) and lithium are widely used in the treatment of bipolar disorder. However, the underlying mechanism of action of these drugs is not clearly understood. We used RNA-Seq analysis to examine the global profile of gene expression in a rat serotonergic cell line (RN46A) after exposure to these two mood stabilizer drugs. Numerous genes were differentially regulated in response to VPA (log2 fold change ≥ 1.0; i.e. -
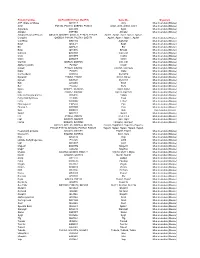
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

High-Fidelity CRISPR/Cas9
ARTICLE DOI: 10.1038/s41467-018-05766-5 OPEN High-fidelity CRISPR/Cas9- based gene-specific hydroxymethylation rescues gene expression and attenuates renal fibrosis Xingbo Xu1,2, Xiaoying Tan2,3, Björn Tampe 3, Tim Wilhelmi1,2, Melanie S. Hulshoff1,2,4, Shoji Saito3, Tobias Moser 5, Raghu Kalluri6, Gerd Hasenfuss1,2, Elisabeth M. Zeisberg1,2 & Michael Zeisberg2,3 fi 1234567890():,; While suppression of speci c genes through aberrant promoter methylation contributes to different diseases including organ fibrosis, gene-specific reactivation technology is not yet available for therapy. TET enzymes catalyze hydroxymethylation of methylated DNA, reac- tivating gene expression. We here report generation of a high-fidelity CRISPR/Cas9-based gene-specific dioxygenase by fusing an endonuclease deactivated high-fidelity Cas9 (dHFCas9) to TET3 catalytic domain (TET3CD), targeted to specific genes by guiding RNAs (sgRNA). We demonstrate use of this technology in four different anti-fibrotic genes in different cell types in vitro, among them RASAL1 and Klotho, both hypermethylated in kidney fibrosis. Furthermore, in vivo lentiviral delivery of the Rasal1-targeted fusion protein to interstitial cells and of the Klotho-targeted fusion protein to tubular epithelial cells each results in specific gene reactivation and attenuation of fibrosis, providing gene-specific demethylating technology in a disease model. 1 Department of Cardiology and Pneumology, University Medical Center Göttingen, Robert-Koch-Str. 40, 37075 Göttingen, Germany. 2 German Center for Cardiovascular Research (DZHK) Partner Site, Göttingen, Germany. 3 Department of Nephrology and Rheumatology, University Medical Center Göttingen, Robert-Koch-Str. 40, 37075 Göttingen, Germany. 4 Department of Pathology and Medical Biology, University Medical Center Groningen, Hanzeplein 1, 9713 Groningen, GZ, Netherlands.