Genetic and Functional Studies of Hirschsprung Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Stelios Pavlidis3, Matthew Loza3, Fred Baribaud3, Anthony
Supplementary Data Th2 and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in UBIOPRED Chih-Hsi Scott Kuo1.2, Stelios Pavlidis3, Matthew Loza3, Fred Baribaud3, Anthony Rowe3, Iaonnis Pandis2, Ana Sousa4, Julie Corfield5, Ratko Djukanovic6, Rene 7 7 8 2 1† Lutter , Peter J. Sterk , Charles Auffray , Yike Guo , Ian M. Adcock & Kian Fan 1†* # Chung on behalf of the U-BIOPRED consortium project team 1Airways Disease, National Heart & Lung Institute, Imperial College London, & Biomedical Research Unit, Biomedical Research Unit, Royal Brompton & Harefield NHS Trust, London, United Kingdom; 2Department of Computing & Data Science Institute, Imperial College London, United Kingdom; 3Janssen Research and Development, High Wycombe, Buckinghamshire, United Kingdom; 4Respiratory Therapeutic Unit, GSK, Stockley Park, United Kingdom; 5AstraZeneca R&D Molndal, Sweden and Areteva R&D, Nottingham, United Kingdom; 6Faculty of Medicine, Southampton University, Southampton, United Kingdom; 7Faculty of Medicine, University of Amsterdam, Amsterdam, Netherlands; 8European Institute for Systems Biology and Medicine, CNRS-ENS-UCBL, Université de Lyon, France. †Contributed equally #Consortium project team members are listed under Supplementary 1 Materials *To whom correspondence should be addressed: [email protected] 2 List of the U-BIOPRED Consortium project team members Uruj Hoda & Christos Rossios, Airways Disease, National Heart & Lung Institute, Imperial College London, UK & Biomedical Research Unit, Biomedical Research Unit, Royal -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
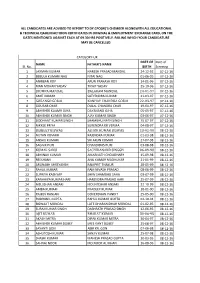
Sl. No. NAME FATHER's NAME DATE of BIRTH Date of Screeing 1 LAXMAN KUMAR NARESH PRASAD MANDAL 24-12-92 07-12-16 2 BEBULA KUMARI
ALL CANDIDATES ARE ADVISED TO REPORT TO DY.CPO(W)'S CHEMBER ALONGWITH ALL EDUCATIONAL & TECHNICAL QAULIFIIACTIOON CERTIFICATES IN ORIGINAL & EMJPLOYMENT EXCHANGE CARD, ON THE DATES MENTIONED AGAINST EACH AT 09.30 HRS POSITIVELY. FAILING WHICH YOUR CANDIDATURE MAY BE CANCELLED. CATEGORY-UR DATE OF Date of NAME FATHER'S NAME Sl. No. BIRTH Screeing 1 LAXMAN KUMAR NARESH PRASAD MANDAL 24-12-92 07-12-16 2 BEBULA KUMARI NAG NITAI NAG 01-06-95 07-12-16 3 ANIRBAN ROY ARUN PRAKASH ROY 14-01-96 07-12-16 4 MAN MOHAN YADAV TIKAIT YADAV 25-10-96 07-12-16 5 JHUMPA MANDAL BALARAM MANDAL 24-01-97 07-12-16 6 AMIT KUMAR SATYENDRA KUMAR 21-03-97 07-12-16 7 GORANGO GORAI KANHAY CHANDRA GORAI 22-03-97 07-12-16 8 GOURAB CHAR DULAL CHANDRA CHAR 19-04-97 07-12-16 9 ABHISHEK KUAMR OJHA DAYANAND OJHA 05-05-97 07-12-16 10 ABHISHEK KUMAR SINGH AJAY KUMAR SINGH 03-06-97 07-12-16 11 SIDDHANT KUMAR SINGH SHAMBHU NATH SINGH 15-07-97 07-12-16 12 NIKKEE PRIYA SURENDRA KR VERMA 04-08-97 07-12-16 13 DEBJEET BISWAS ASHOK KUMAR BISWAS 13-02-98 08-12-16 14 NUTAN KUMARI RAJENDRA KUMAR 01-03-98 08-12-16 15 ANSHU KUMARI SRI ARUN KUMAR 27-07-98 08-12-16 16 SAGNIK PURI CHANDIDAS PURI 03-08-98 08-12-16 17 KUMAR SANU SACHIDANAND SINGGH 06-09-98 08-12-16 18 ABHINAV KUMAR AMARNATH CHOUDHARY 15-09-98 08-12-16 19 REESHANK ANIL KUMAR MADHUKAR 21-01-99 08-12-16 20 ARUNABH SHREYANSH RAMPRIT THAKUR 28-05-99 08-12-16 21 RAHUL KUMAR RAM NIVASH PRASAD 08-06-99 08-12-16 22 SUPRIYA KASHYAP SHIW SHAMBHU SAHA 03-07-99 08-12-16 23 KANHAIYA KUMAR HARI HARENDRA PRASAD HARI 25-07-99 08-12-16 24 MD.JISHAN ANSARI MD.INTISHAR ANSARI 22-10-99 08-12-16 25 AMBA KUMARI PRADEEP KUMAR 18-01-00 08-12-16 26 RAJEEV RANJAN GOVERDHAN PANDIT 25-05-00 08-12-16 27 RUDRANIL GUPTA RAHUL KUMAR GUPTA 02-10-94 08-12-16 28 BISWAJIT MONDAL LATE DHARANIDHAR MONDAL 01-02-95 08-12-16 29 SURAJ KUMAR SINGH DASHRATH PRASAD SINGH 12-06-95 08-12-16 30 AJIT KUMAR BHARAT KUMAR 30-06-95 08-12-16 31 AKASH MITRA DIPAK KUMAR MITRA 30-04-97 08-12-16 32 ABHISHEK KUMAR DUBEY SREE KANT DUBEY 25-08-97 08-12-16 33 SAYANTI GON ABIR KR. -

Macropinocytosis Requires Gal-3 in a Subset of Patient-Derived Glioblastoma Stem Cells
ARTICLE https://doi.org/10.1038/s42003-021-02258-z OPEN Macropinocytosis requires Gal-3 in a subset of patient-derived glioblastoma stem cells Laetitia Seguin1,8, Soline Odouard2,8, Francesca Corlazzoli 2,8, Sarah Al Haddad2, Laurine Moindrot2, Marta Calvo Tardón3, Mayra Yebra4, Alexey Koval5, Eliana Marinari2, Viviane Bes3, Alexandre Guérin 6, Mathilde Allard2, Sten Ilmjärv6, Vladimir L. Katanaev 5, Paul R. Walker3, Karl-Heinz Krause6, Valérie Dutoit2, ✉ Jann N. Sarkaria 7, Pierre-Yves Dietrich2 & Érika Cosset 2 Recently, we involved the carbohydrate-binding protein Galectin-3 (Gal-3) as a druggable target for KRAS-mutant-addicted lung and pancreatic cancers. Here, using glioblastoma patient-derived stem cells (GSCs), we identify and characterize a subset of Gal-3high glio- 1234567890():,; blastoma (GBM) tumors mainly within the mesenchymal subtype that are addicted to Gal-3- mediated macropinocytosis. Using both genetic and pharmacologic inhibition of Gal-3, we showed a significant decrease of GSC macropinocytosis activity, cell survival and invasion, in vitro and in vivo. Mechanistically, we demonstrate that Gal-3 binds to RAB10, a member of the RAS superfamily of small GTPases, and β1 integrin, which are both required for macro- pinocytosis activity and cell survival. Finally, by defining a Gal-3/macropinocytosis molecular signature, we could predict sensitivity to this dependency pathway and provide proof-of- principle for innovative therapeutic strategies to exploit this Achilles’ heel for a significant and unique subset of GBM patients. 1 University Côte d’Azur, CNRS UMR7284, INSERM U1081, Institute for Research on Cancer and Aging (IRCAN), Nice, France. 2 Laboratory of Tumor Immunology, Department of Oncology, Center for Translational Research in Onco-Hematology, Swiss Cancer Center Léman (SCCL), Geneva University Hospitals, University of Geneva, Geneva, Switzerland. -

Hirschsprung´S Disease & Gastroesophageal Reflux. Aspects on Two Gastrointestinal Motility Disorders in Childhood Gunnarsdo
Hirschsprung´s Disease & Gastroesophageal Reflux. Aspects on Two Gastrointestinal Motility Disorders in Childhood Gunnarsdottir, Anna 2010 Link to publication Citation for published version (APA): Gunnarsdottir, A. (2010). Hirschsprung´s Disease & Gastroesophageal Reflux. Aspects on Two Gastrointestinal Motility Disorders in Childhood. Lund University: Faculty of Medicine. Total number of authors: 1 General rights Unless other specific re-use rights are stated the following general rights apply: Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal Read more about Creative commons licenses: https://creativecommons.org/licenses/ Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. LUND UNIVERSITY PO Box 117 221 00 Lund +46 46-222 00 00 Hirschsprung´s Disease & Gastroesophageal Reflux Aspects on Two Gastrointestinal Motility Disorders in Childhood Akademisk avhandling I ämnet -

Systematic Proteome and Proteostasis Profiling in Human Trisomy
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Open Repository and Bibliography - Luxembourg ARTICLE DOI: 10.1038/s41467-017-01422-6 OPEN Systematic proteome and proteostasis profiling in human Trisomy 21 fibroblast cells Yansheng Liu 1, Christelle Borel2,LiLi3, Torsten Müller1, Evan G. Williams1, Pierre-Luc Germain4, Marija Buljan1, Tatjana Sajic1, Paul J. Boersema5, Wenguang Shao1, Marco Faini1, Giuseppe Testa4,6, Andreas Beyer 3, Stylianos E. Antonarakis2,7 & Ruedi Aebersold 1,8 Down syndrome (DS) is mostly caused by a trisomy of the entire Chromosome 21 (Trisomy 21, T21). Here, we use SWATH mass spectrometry to quantify protein abundance and protein turnover in fibroblasts from a monozygotic twin pair discordant for T21, and to profile protein expression in 11 unrelated DS individuals and matched controls. The integration of the steady- state and turnover proteomic data indicates that protein-specific degradation of members of stoichiometric complexes is a major determinant of T21 gene dosage outcome, both within and between individuals. This effect is not apparent from genomic and transcriptomic data. The data also reveal that T21 results in extensive proteome remodeling, affecting proteins encoded by all chromosomes. Finally, we find broad, organelle-specific post-transcriptional effects such as significant downregulation of the mitochondrial proteome contributing to T21 hallmarks. Overall, we provide a valuable proteomic resource to understand the origin of DS phenotypic manifestations. 1 Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, 8093 Zurich, Switzerland. 2 Department of Genetic Medicine and Development, University of Geneva Medical School, and University Hospitals of Geneva, 1211 Geneva, Switzerland. -
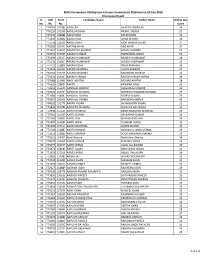
Sl. No. Roll No. Form No. Candidate Name Father Name Online Test
RUHS Paramedical UG/Diploma Entrance Examination 2018 held on 22-July-2018 Provisional Result Sl. Roll Form Candidate Name Father Name Online test No. No. No. Score 1 774709 157580 AADIL ALI AKHTAR JAWED ALI 24 2 776110 152356 AADIL HUSSAIN MOHD. SADDIK 23 3 775335 158688 AADIL KHAN ZAFAR KHAN 30 4 773345 153862 AADIL KHAN ILIYAS AHMED 34 5 775548 158239 AADIL SHEKH MOH ANWAR SHEKH 27 6 770300 150497 AAFTAB KHAN AZIZ KHAN 27 7 771637 154595 AAKANSHA SHARMA KAMAL SHARMA 27 8 774515 157095 AAKASH KUMAR MAHENDRA SINGH 53 9 775699 150357 AAKASH KUMAWAT MUKESH KUMAWAT 28 10 775130 156807 AAKASH KUMAWAT SURESH KUMAWAT 31 11 774711 152808 AAKASH OJHA PREM PRAKASH 33 12 774032 155356 AAKASH SHARMA LALITA SHARMA 19 13 774263 157776 AAKASH SHARMA RAMDHAN SHARMA 20 14 775634 156007 AAKASH VERMA RAJESH KUMAR VERMA 28 15 773588 151461 AAKIF AKHTAR ARSHAD AKHTAR 28 16 776626 158824 AAKRTI KANWAR SINGH 26 17 770659 155640 AANCHAL DINDOR MAGANLAL DINDOR 22 18 775121 154734 AANCHAL SHARMA MAHESH CHANDRA SHARMA 24 19 772086 158087 AANCHAL SUMAN SURESH SUMAN 25 20 775097 150908 AANCHAL VERMA RAJENDRA VERMA 49 21 774632 152376 AARAV YADAV JAI NARAYAN YADAV 23 22 771846 157780 AAROHEE SHARMA Kishan lal loknathaka 30 23 772930 155169 AARTI DHABHAI BADRI NARAYAN DHABHAI 29 24 770741 151563 AARTI GURJAR DEVKARAN GURJAR 24 25 773160 156830 AARTI JAIN BHAGCHAND JAIN 34 26 773407 154289 AARTI JAIPAL TEJARAM JAIPAL 32 27 771420 157114 AARTI MEGHWAL ASHOK KUMAR 31 28 772749 152887 AARTI PANWAR SUBHASH CHAND VERMA 19 29 775073 153889 AARTI SHARMA ROOP NARAYAN SHARMA -

Movies of Feroz Khan
1 / 2 Movies Of Feroz Khan Feroze Khan (born July 11, 1990) is famous for being tv actor. ... Padmavati is undeniably the most awaited movie of the year and it won't be hard to call it a hit .... Feroz Khan Movies List. Thambi Arjuna (2010)Ramana, Aashima. Welcome (2007)Feroz Khan, Anil Kapoor. Ek Khiladi Ek Haseena (2005)Fardeen Khan, .... See more ideas about actors, pakistani actress, feroz khan. ... Khushbo is Pakistani film and stage Actress. c) On some minimal productions (e. Want to Become .... Tasveer Full Hindi Movie (1966) Film cast : Feroz Khan, Kalpana, Helen, Sajjan, Rajendra Nath, Nazir Hasain, Leela Mishra, Raj Mehra, Lalita Pawar, Sabeena. MUMBAI, India (Agence France-Presse) — Feroz Khan, a Bollywood ... he made “Dharmatma,” the first Hindi-language movie shot on location .... Actor Rishi Kapoor tweeted that the two actors, who worked together in three films, died of Cancer on the same date. Known for his flamboyant persona, Feroz Khan was an actor, director and producer who had a long innings in the Hindi film industry. The actor .... Feroz Khan was born at Bangalore in India to an Afghan Father, Sadiq Khan and an Iranian mother Fatima. After schooling from Bangalore he shifted to Bombay .... He would have turned 79 on September 25. However, actor, director and producer Feroz Khan – one of his kind in the Hindi film industry .... Feroz Khan All Movies List · Ek Khiladi Ek Haseena - 2005 · Ek Khiladi Ek Haseena (2005)Feroz Khan , Fardeen Khan , Koena Mitra , Gulshan Grover , Kay Kay , .... India's answer to Clint Eastwood, Feroz Khan is known as the most stylish bollywood actor. -

Early Growth Response 1 Regulates Hematopoietic Support and Proliferation in Human Primary Bone Marrow Stromal Cells
Hematopoiesis SUPPLEMENTARY APPENDIX Early growth response 1 regulates hematopoietic support and proliferation in human primary bone marrow stromal cells Hongzhe Li, 1,2 Hooi-Ching Lim, 1,2 Dimitra Zacharaki, 1,2 Xiaojie Xian, 2,3 Keane J.G. Kenswil, 4 Sandro Bräunig, 1,2 Marc H.G.P. Raaijmakers, 4 Niels-Bjarne Woods, 2,3 Jenny Hansson, 1,2 and Stefan Scheding 1,2,5 1Division of Molecular Hematology, Department of Laboratory Medicine, Lund University, Lund, Sweden; 2Lund Stem Cell Center, Depart - ment of Laboratory Medicine, Lund University, Lund, Sweden; 3Division of Molecular Medicine and Gene Therapy, Department of Labora - tory Medicine, Lund University, Lund, Sweden; 4Department of Hematology, Erasmus MC Cancer Institute, Rotterdam, the Netherlands and 5Department of Hematology, Skåne University Hospital Lund, Skåne, Sweden ©2020 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2019.216648 Received: January 14, 2019. Accepted: July 19, 2019. Pre-published: August 1, 2019. Correspondence: STEFAN SCHEDING - [email protected] Li et al.: Supplemental data 1. Supplemental Materials and Methods BM-MNC isolation Bone marrow mononuclear cells (BM-MNC) from BM aspiration samples were isolated by density gradient centrifugation (LSM 1077 Lymphocyte, PAA, Pasching, Austria) either with or without prior incubation with RosetteSep Human Mesenchymal Stem Cell Enrichment Cocktail (STEMCELL Technologies, Vancouver, Canada) for lineage depletion (CD3, CD14, CD19, CD38, CD66b, glycophorin A). BM-MNCs from fetal long bones and adult hip bones were isolated as reported previously 1 by gently crushing bones (femora, tibiae, fibulae, humeri, radii and ulna) in PBS+0.5% FCS subsequent passing of the cell suspension through a 40-µm filter. -

Shankar Jaikishan – Emperors of Music by Ashwini Kumar Rath
Shankar Jaikishan – Emperors of Music By Ashwini Kumar Rath Shankar Jaikishan music, synonym of Indian Film music, has far reaching impact in India and outside. The duo moulded music and musical mood of masses since 1949, and are still considered to be the best music composers from India. The duo also contributed outside movie industry with their much celebrated album Raga in Jazz Style, and background music for documentary Everest expedition among many others. If we look at the popular impact by any personalities in the field of music during the post-independence period in India, the obvious choice is Shankar Jaikishan. Shankar, the more versatile of the two, was a prolific musician with mastery over dozens of instruments, Indian classical music and dance forms. He was primarily responsible for building Shankar Jaikishan team with dedicated lyricists, music assistants, and jumbo-sized 60-pieces orchestra, and was the lead composer. On the other hand, Jaikishan was incredibly creative and legendary composer himself. He was genius of background music and romantic tunes though he too could compose songs in other genres. While Shankar's musical jhankar propels divine dwellers to zoom in unison with ecstasy, romantic effervescence of Jaikishan music brings divine feelings on to earth. During initial years, all decisions about musical engagements were made by Shankar; but both started taking up assignments separately in the late sixties. They had sharp contrasts in personalities and physical appearances. Shankar was gym-savvy, non- drinker, and was a strict disciplinarian. On the other hand, Jaikishan was party-man and was very social. -

Download Registration Number for Offline
To download the Admit Card please enter your Registration Number and Date of Birth in the format (yyyy-mm-dd). Registration Number Name of Candidate Course1 Course2 Father Name CUJET/2012/000001 JAI SHREE BHARDWAJ M.Tech. Nano Technology M.Tech. Energy Engineering KUMAR SUNDRAM BHARDWAJ CUJET/2012/000002 DHRAMENDAR KUMAR M.B.A. M.A. International Relations Harinath CUJET/2012/000003 KAJOL M.Tech. Nano Technology M.Tech. Energy Engineering SUMAN KUMAR VERMA CUJET/2012/000004 Ashutosh Kumar M.Tech. Energy Engineering M.Tech. Water Engineering & Management Madan Murari Pandey CUJET/2012/000005 RAVI VIKASH TIRKEY M.B.A. M.A. Mass Communication LEO ABHAY TIRKEY CUJET/2012/000006 Ms. SAYANTANI MANDAL M.Sc. Environmental Science Mr. NISITH RANJAN MANDAL CUJET/2012/000012 Mr OMKAR SHARAN MISHRA M.B.A. M.A. International Relations Mr KAMAL KUMAR MISHRA Mr ABHIRAM SHARAN CUJET/2012/000013 MISHRA M.Tech. Energy Engineering M.Sc. Applied Chemistry Mr KAMAL KUMAR MISHRA CUJET/2012/000014 Samatha Tejavath M.Sc. Applied Mathematics M.Tech. Nano Technology Nageswara Rao Tejavath CUJET/2012/000015 ASHUTOSH GUPTA M.Tech. Energy Engineering M.Tech. Water Engineering & Management A K GUPTA CUJET/2012/000017 Shahid Hussain Rahmatullah CUJET/2012/000019 RAJIV RANJAN M.B.A. M.A. Mass Communication SURYADEO SINGH CUJET/2012/000020 Amit Kumar M.B.A. M.A. Mass Communication Rahul Kumar CUJET/2012/000021 arman khan M.Tech. Nano Technology M.Tech. Water Engineering & Management arif khan CUJET/2012/000023 AMIT ANAND M.B.A. M.Tech. Nano Technology VEER PRASAD MAHTO CUJET/2012/000024 MOHIT MAYOOR M.Tech. -

Structural Study of the Acid Sphingomyelinase Protein Family
Structural Study of the Acid Sphingomyelinase Protein Family Alexei Gorelik Department of Biochemistry McGill University, Montreal August 2017 A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Doctor of Philosophy © Alexei Gorelik, 2017 Abstract The acid sphingomyelinase (ASMase) converts the lipid sphingomyelin (SM) to ceramide. This protein participates in lysosomal lipid metabolism and plays an additional role in signal transduction at the cell surface by cleaving the abundant SM to ceramide, thus modulating membrane properties. These functions are enabled by the enzyme’s lipid- and membrane- interacting saposin domain. ASMase is part of a small family along with the poorly characterized ASMase-like phosphodiesterases 3A and 3B (SMPDL3A,B). SMPDL3A does not hydrolyze SM but degrades extracellular nucleotides, and is potentially involved in purinergic signaling. SMPDL3B is a regulator of the innate immune response and podocyte function, and displays a partially defined lipid- and membrane-modifying activity. I carried out structural studies to gain insight into substrate recognition and molecular functions of the ASMase family of proteins. Crystal structures of SMPDL3A uncovered the helical fold of a novel C-terminal subdomain, a slightly distinct catalytic mechanism, and a nucleotide-binding mode without specific contacts to their nucleoside moiety. The ASMase investigation revealed a conformational flexibility of its saposin domain: this module can switch from a detached, closed conformation to an open form which establishes a hydrophobic interface to the catalytic domain. This open configuration represents the active form of the enzyme, likely allowing lipid access to the active site. The SMPDL3B structure showed a narrow, boot-shaped substrate binding site that accommodates the head group of SM.