Dashboard-Style Interactive Plots for RNA-Seq Analysis Are R Markdown Ready with Glimma 2.0
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Advanced R Second Edition Chapman & Hall/CRC the R Series Series Editors John M
Advanced R Second Edition Chapman & Hall/CRC The R Series Series Editors John M. Chambers, Department of Statistics, Stanford University, California, USA Torsten Hothorn, Division of Biostatistics, University of Zurich, Switzerland Duncan Temple Lang, Department of Statistics, University of California, Davis, USA Hadley Wickham, RStudio, Boston, Massachusetts, USA Recently Published Titles Testing R Code Richard Cotton R Primer, Second Edition Claus Thorn Ekstrøm Flexible Regression and Smoothing: Using GAMLSS in R Mikis D. Stasinopoulos, Robert A. Rigby, Gillian Z. Heller, Vlasios Voudouris, and Fernanda De Bastiani The Essentials of Data Science: Knowledge Discovery Using R Graham J. Williams blogdown: Creating Websites with R Markdown Yihui Xie, Alison Presmanes Hill, Amber Thomas Handbook of Educational Measurement and Psychometrics Using R Christopher D. Desjardins, Okan Bulut Displaying Time Series, Spatial, and Space-Time Data with R, Second Edition Oscar Perpinan Lamigueiro Reproducible Finance with R Jonathan K. Regenstein, Jr. R Markdown The Definitive Guide Yihui Xie, J.J. Allaire, Garrett Grolemund Practical R for Mass Communication and Journalism Sharon Machlis Analyzing Baseball Data with R, Second Edition Max Marchi, Jim Albert, Benjamin S. Baumer Spatio-Temporal Statistics with R Christopher K. Wikle, Andrew Zammit-Mangion, and Noel Cressie Statistical Computing with R, Second Edition Maria L. Rizzo Geocomputation with R Robin Lovelace, Jakub Nowosad, Jannes Münchow Dose-Response Analysis with R Christian Ritz, Signe M. Jensen, Daniel Gerhard, Jens C. Streibig Advanced R, Second Edition Hadley Wickham For more information about this series, please visit: https://www.crcpress.com/go/ the-r-series Advanced R Second Edition Hadley Wickham CRC Press Taylor & Francis Group 6000 Broken Sound Parkway NW, Suite 300 Boca Raton, FL 33487-2742 © 2019 by Taylor & Francis Group, LLC CRC Press is an imprint of Taylor & Francis Group, an Informa business No claim to original U.S. -

Data Analysis in R
Data Analysis in R Course at a Glance This course will provide an introduction to reproducible data analysis with R (see Syllabus). Instructor Gabriel Baud-Bovy ([email protected] ) Credits: 5 Synopsis This course aims at giving to the student a methodology to analyze experimental results, from how to organize data to the writing of a report. It includes: an introduction to R an introduction to reproducible research with R examples of statistical analysis with R During this course, the student will have to analyze his own data and is expected to read before each course the material that will be made available on this page. The final grade will consist in the evaluation of a report demonstrating familiarity with the concepts and methods presented in the course. As an editor, the instructor will use Notepad++ (together with NppToR) on a Windows Machines but the student might use other ones (e.g., R studio, EMACS+ESS, Lyx, TexWork). The course will use Mardown as typesetting language. For those desirous to work with Latex and/or generate pdf, you will need to install also MikeTex. Syllabus Total of 15 hours Class 1: Case study, Reproducible research Class 2: R fundamental Class 3: Exploratory data analysis and graphical methods in R Class 4: Basic statistics Class 5: To be determined There will be a final examination decided by the instructor. Prerequisites The course assumes some familiarity with programming concepts and data structures (MATLAB, C/C++, Java or any other programming language). Contact the instructor if you have never programmed anything. -

Language-Agnostic Data Analysis Workflows and Reproducible Research
Language-agnostic data analysis workflows and reproducible research Andrew John Lowe 28 April 2017 Abstract This is the abstract for a document that briefly describes methods for interacting with different programming languages within the same data analysis workflow. 0.1 Overview • This talk: language-agnostic (or polyglot) analysis workflows • I’ll show how it is possible to work in multiple languages and switch between them without leaving the workflow you started • Additionally, I’ll demonstrate how an entire workflow can be encapsulated in a markdown file that is rendered to a publishable paper with cross-references and a bibliography (and with raw a LATEX file produced as a by-product) in a simple process, making the whole analysis workflow reproducible 0.2 Which tool/language is best? • TMVA, scikit-learn, h2o, caret, mlr, WEKA, Shogun, . • C++, Python, R, Java, MATLAB/Octave, Julia, . • Many languages, environments and tools • People may have strong opinions • A false polychotomy? • Be a polyglot! 0.3 Approaches • Save-and-load • Language-specific bindings • Notebook • knitr 1 Save-and-load approach 1.1 Flat files • Language agnostic • Formats like text, CSV, and JSON are well-supported • Breaks workflow • Data types may not be preserved (e.g., datetime, NULL) • New binary format Feather solves this 1 1.2 Feather Feather: A fast on-disk format for data frames for R and Python, powered by Apache Arrow, developed by Wes McKinney and Hadley Wickham # In R: library(feather) path <- "my_data.feather" write_feather(df, path) # In Python: import feather -
Dynamic Documents with R and Knitr, by Yihui Xie 116 Tugboat, Volume 35 (2014), No
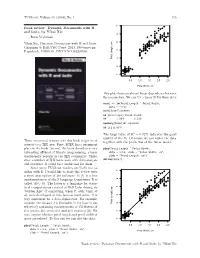
TUGboat, Volume 35 (2014), No. 1 115 ● ● ● Book review: Dynamic Documents with R ● ● ● and knitr, by Yihui Xie ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● Boris Veytsman ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● Yihui Xie, Dynamic Documents with R and knitr. ● ● ● ● ● ● ● ● ● ● ● ● ● ● Chapman & Hall/CRC Press, 2013, 190+xxvi pp. ● ● ● ● ● ● ● ● ● US$ ISBN ● Paperback, 59.95. 978-1482203530. ● ● ● ● Petal Length, cm Petal ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 1 2 3 4 5 6 7 0.5 1.0 1.5 2.0 2.5 Petal Width, cm This plot shows an almost linear dependence between the parameters. We can try a linear fit for these data: model <- lm(Petal.Length ˜ Petal.Width, data = iris) model$coefficients ## (Intercept) Petal.Width ## 1.084 2.230 summary(model)$r.squared ## [1] 0.9271 The large value of R2 = 0.9271 indicates the good quality of the fit. Of course we can replot the data There are several reasons why this book might be of together with the prediction of the linear model: interest to a TEX user. First, LATEX has a prominent place in the book. Second, the book describes a very plot(Petal.Length ˜ Petal.Width, interesting offshoot of literate programming, a topic data = iris, xlab = "Petal Width, cm", traditionally popular in the TEX community. Third, ylab = "Petal Length, cm") abline(model) since a number of TEX users work with data analysis and statistics, R could be a useful tool for them. Since some TUGboat readers are likely not fa- ● ● ● miliar with R, I would like to start this review with ● ● a short description of the software. R [1] is a free ● ● ● ● ● ● ● ● ● ● ● implementation of the S language (sometimes R is ● ● ● ● ● ● ● ● ● ● ● ● ● ● called GNU S). -

LYX and Knitr
knitr.1 LYX and knitr The knitr package allows one to embed R code within LYX and LATEX documents. When a document is compiled into a PDF, LYX/LATEX connects to R to run the R code and the code/output is automatically put into the PDF. In addition to this being a convenient way to use both LYX/LATEX with R, it also provides an important component to the reproducibility of research (RR). For example, one can include the code for a data analysis de- scribed in a paper. This ensures that there would be no “copying and pasting errors” and also provide readers of the paper an im- mediate way to reproduce the research. RR continues to become more important and fortunately more tools are being developed to make it possible. Below are some discussions on the topic: • AMSTAT News column on RR at http://magazine. amstat.org/blog/2011/01/01/scipolicyjan11. • CRAN task view for RR and R at http://cran.r-project. org/web/views/ReproducibleResearch.html. • Yihui Xie: Author of knitr – First and second editions of his Dynamic Documents with R and knitr book. Note that this book was typed in LYX. – Website for knitr at http://yihui.name/knitr The Sweave environment is another way to include R code inside of LYX/LATEX. This was developed prior to knitr, but it is more difficult to use. The purpose of this section is to examine the main compo- nents of knitr so that you will be able to complete the rest of the semester using LYX and knitr together for all assign- ments in our course! Also, a very important purpose is to give you the tools needed to complete all assignments in other R- based courses by using knitr and LYX together! The files used knitr.2 here are intro_example_cereal.lyx, intro_example_cereal.pdf, cereal.csv, ExternalCode.R, FirstBeamer-knitr.zip, JSM2015.zip, and RMarkdown.zip. -

The R Journal, June 2012
The Journal Volume 2/1, June 2010 A peer-reviewed, open-access publication of the R Foundation for Statistical Computing Contents Editorial..................................................3 Contributed Research Articles IsoGene: An R Package for Analyzing Dose-response Studies in Microarray Experiments..5 MCMC for Generalized Linear Mixed Models with glmmBUGS ................. 13 Mapping and Measuring Country Shapes............................... 18 tmvtnorm: A Package for the Truncated Multivariate Normal Distribution........... 25 neuralnet: Training of Neural Networks............................... 30 glmperm: A Permutation of Regressor Residuals Test for Inference in Generalized Linear Models.................................................. 39 Online Reproducible Research: An Application to Multivariate Analysis of Bacterial DNA Fingerprint Data............................................ 44 Two-sided Exact Tests and Matching Confidence Intervals for Discrete Data.......... 53 Book Reviews A Beginner’s Guide to R......................................... 59 News and Notes Conference Review: The 2nd Chinese R Conference........................ 60 Introducing NppToR: R Interaction for Notepad++......................... 62 Changes in R 2.10.1–2.11.1........................................ 64 Changes on CRAN............................................ 72 News from the Bioconductor Project.................................. 85 R Foundation News........................................... 86 2 The Journal is a peer-reviewed publication of -

R Markdown: the Definitive Guide
Yihui Xie, J. J. Allaire, Garrett Grolemund R Markdown: The Definitive Guide To Jung Jae-sung (1982 – 2018), a remarkably hard-working badminton player with a remarkably simple playing style Contents List of Tables xvii List of Figures xix Preface xxiii About the Authors xxxv I GetStarted 1 1 Installation 5 2 Basics 7 2.1 Example applications .................. 7 2.1.1 Airbnb’s knowledge repository ......... 7 2.1.2 Homework assignments on RPubs ....... 7 2.1.3 Personalized mail ................. 8 2.1.4 2017 Employer Health Benefits Survey ..... 8 2.1.5 Journal articles .................. 8 2.1.6 Dashboards at eelloo ............... 8 2.1.7 Books ....................... 8 2.1.8 Websites ...................... 8 2.2 Compile an R Markdown document .......... 8 2.3 Cheat sheets ....................... 9 2.4 Output formats ...................... 9 2.5 Markdown syntax .................... 9 2.5.1 Inline formatting ................. 9 2.5.2 Block-level elements ............... 9 2.5.3 Math expressions ................. 9 2.6 R code chunks and inline R code ............ 10 2.6.1 Figures ....................... 10 2.6.2 Tables ....................... 10 vii viii Contents 2.7 Other language engines ................. 10 2.7.1 Python ....................... 10 2.7.2 Shell scripts .................... 10 2.7.3 SQL ........................ 11 2.7.4 Rcpp ........................ 11 2.7.5 Stan ........................ 11 2.7.6 JavaScript and CSS ................ 11 2.7.7 Julia ........................ 11 2.7.8 CandFortran ................... 11 2.8 Interactive documents .................. 11 2.8.1 HTML widgets .................. 11 2.8.2 Shiny documents ................. 12 II Output Formats 13 3 Documents 17 3.1 HTML document ..................... 17 3.1.1 Table of contents ................ -

Stat 302 Statistical Software and Its Applications the Knitr Package and Rstudio
Stat 302 Statistical Software and Its Applications The knitr Package and RStudio Fritz Scholz Department of Statistics, University of Washington Winter Quarter 2015 January 8, 2015 1 The knitr Package The knitr package integrates code, output, and narrative. It was created by Yihui Xie who was inspired by Sweave. Right click on the R icon and start an Administrator R session. Install knitr via install.packages("knitr", dependencies=TRUE) It is assumed that LATEX is already installed on your system. In the initial use of knitr in R or RStudio you may be prompted to install further LATEX packages. That happens just once and is automatic. 2 First Usage of knitr Create a working directory, say EX2. Inside EX2 and using a text editor create a le named minimal.Rnw with content shown on the next two slides. Note that it looks very much like a LATEX le in structure, except for the chunk delimited by <<plot .... >>= R code commands @ Also note the use of \Sexpr{...} to access contents of R objects created with the R commands. You can use ’right’ or ’left’ in place of ’center’ in fig.align. The plot after << acts as plot le name handle, see the gure subfolder in EX2. Dierent plots require dierent handles. 3 Content of minimal.Rnw (Slide 1) \documentclass{article} \setlength{\topmargin}{-1in} \setlength{\oddsidemargin}{-.25in} \setlength{\evensidemargin}{-.25in} \setlength{\textwidth}{7in} \setlength{\textheight}{10in} \begin{document} \begin{flushright} \parbox{1in}{Fritz Scholz\\ ID: 1234567\vspace{1.25in} } \includegraphics[width=.5in]{./figure/fritzbuzz.jpg} \end{flushright} \begin{center} {\large A Minimal Example}\vspace{.1in}\\ Fritz Scholz \end{center} We examine the relationship between speed and stopping distance using a linear regression model: $Y=\beta_0+\beta_1 x+\epsilon$. -

Reproducible Research Tools for R
Reproducible Research Tools for R January 2017 Boriana Pratt Princeton University 1 Literate programming Literate programming (1984) “ I believe that the time is right for significantly better documentation of programs, and that we can best achieve this by considering programs to be works of literature. Hence, my title: ‘Literate Programming’. “ Donald E. Knuth. Literate Programming. The Computer Journal, 27(2):97-111, May1984. http://comjnl.oxfordjournals.org/content/27/2/97.full.pdf+html WEB “… is a combination of two other languages: (1) a document formatting language and (2) a programming language.” 2 terms • Tangle extract the code parts (code chunks), then run them sequentially • Weave extract the text part (documentation chunks) and weave back in the code and code output 3 Noweb Norman Ramsey (1989) – Noweb – simple literate programming tool… https://www.cs.tufts.edu/~nr/noweb/ “Literate programming is the art of preparing programs for human readers. “ Noweb syntax includes two parts: Code chunk: <<chink name>>= - section that starts with <<name>>= Documentation chunk: @ - line that starts with @ followed by a space; default for the first chunk 4 Tools for R 5 Tools for R • Sweave https://stat.ethz.ch/R-manual/R-devel/library/utils/doc/Sweave.pdf What is Sweave? A tool that allows to embed the R code in LaTeX documents. The purpose is to create dynamic reports, which can be updated automatically if data or analysis change. How do I cite Sweave? To cite Sweave please use the paper describing the first version: Friedrich Leisch. Sweave: Dynamic generation of statistical reports using literate data analysis. -

Package 'Ymlthis'
Package ‘ymlthis’ September 15, 2021 Title Write 'YAML' for 'R Markdown', 'bookdown', 'blogdown', and More Version 0.1.5 Description Write 'YAML' front matter for R Markdown and related documents. Work with 'YAML' objects more naturally and write the resulting 'YAML' to your clipboard or to 'YAML' files related to your project. License MIT + file LICENSE URL https://ymlthis.r-lib.org, https://github.com/r-lib/ymlthis BugReports https://github.com/r-lib/ymlthis/issues Depends R (>= 3.2) Imports crayon, fs, glue, magrittr, miniUI, purrr (>= 0.3.2), rlang (>= 0.4.0), rmarkdown (>= 2.10), rstudioapi, shiny, shinyBS, stringr, usethis (>= 1.5.0), whoami, withr, yaml Suggests blogdown, bookdown, covr, knitr, pkgdown, prettydoc, roxygen2 (>= 7.0.0), spelling, testthat, xaringan VignetteBuilder knitr Encoding UTF-8 Language en-US RoxygenNote 7.1.2 NeedsCompilation no Author Malcolm Barrett [aut, cre] (<https://orcid.org/0000-0003-0299-5825>), Richard Iannone [aut] (<https://orcid.org/0000-0003-3925-190X>), RStudio [cph, fnd] Maintainer Malcolm Barrett <[email protected]> Repository CRAN Date/Publication 2021-09-15 08:50:02 UTC 1 2 R topics documented: R topics documented: asis_yaml_output . .3 as_yml . .3 bib2yml . .4 blogdown_template . .5 code_chunk . .5 draw_yml_tree . .6 gitbook_config . .7 has_field . .9 includes2 . 10 is_yml . 10 last_yml . 11 pagedown_business_card_template . 11 pandoc_template_types . 14 pkgdown_template . 15 read_json . 16 use_yml . 17 use_yml_defaults . 18 use_yml_file . 19 yml ............................................. 21 yml_author . 22 yml_blank . 25 yml_blogdown_opts . 25 yml_bookdown_opts . 28 yml_citations . 31 yml_clean . 32 yml_code . 33 yml_distill_opts . 34 yml_handlers . 38 yml_latex_opts . 38 yml_load . 41 yml_output . 42 yml_pagedown_opts . 43 yml_params . 44 yml_pkgdown . 49 yml_reference . 53 yml_replace . 55 yml_resource_files . 56 yml_rsconnect_email . -

Xfun: Supporting Functions for Packages Maintained by 'Yihui Xie'
Package ‘xfun’ September 14, 2021 Type Package Title Supporting Functions for Packages Maintained by 'Yihui Xie' Version 0.26 Description Miscellaneous functions commonly used in other packages maintained by 'Yihui Xie'. Imports stats, tools Suggests testit, parallel, codetools, rstudioapi, tinytex (>= 0.30), mime, markdown, knitr, htmltools, remotes, pak, rhub, renv, curl, jsonlite, rmarkdown License MIT + file LICENSE URL https://github.com/yihui/xfun BugReports https://github.com/yihui/xfun/issues Encoding UTF-8 RoxygenNote 7.1.2 VignetteBuilder knitr NeedsCompilation yes Author Yihui Xie [aut, cre, cph] (<https://orcid.org/0000-0003-0645-5666>), Wush Wu [ctb], Daijiang Li [ctb], Xianying Tan [ctb], Salim Brüggemann [ctb] (<https://orcid.org/0000-0002-5329-5987>), Christophe Dervieux [ctb] Maintainer Yihui Xie <[email protected]> Repository CRAN Date/Publication 2021-09-14 07:10:02 UTC R topics documented: attr..............................................3 base64_encode . .4 base64_uri . .4 1 2 R topics documented: bg_process . .5 broken_packages . .6 bump_version . .6 cache_rds . .7 del_empty_dir . .9 dir_create . .9 dir_exists . 10 download_file . 10 do_once . 11 embed_file . 12 exit_call . 13 file_ext . 14 file_string . 15 format_bytes . 15 from_root . 16 github_releases . 17 grep_sub . 17 gsub_file . 18 install_dir . 19 install_github . 19 in_dir . 20 isFALSE . 20 is_abs_path . 21 is_ascii . 21 is_sub_path . 22 is_web_path . 23 is_windows . 23 magic_path . 24 mark_dirs . 25 msg_cat . 25 native_encode . 26 news2md . 27 normalize_path . 28 numbers_to_words . 28 optipng . 29 parse_only . 30 pkg_attach . 30 process_file . 32 proc_kill . 33 proj_root . 33 prose_index . 34 protect_math . 35 raw_string . 36 read_bin . 37 read_utf8 . 37 relative_path . 38 rename_seq . 39 retry . 40 attr 3 rev_check . 40 Rscript . 43 Rscript_call . 43 rstudio_type . 44 same_path . 45 session_info . 45 set_envvar . 46 split_lines . -

Dissertation Submitted in Partial Satisfaction of the Requirements for the Degree Doctor of Philosophy in Statistics
University of California Los Angeles Bridging the Gap Between Tools for Learning and for Doing Statistics A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Statistics by Amelia Ahlers McNamara 2015 c Copyright by Amelia Ahlers McNamara 2015 Abstract of the Dissertation Bridging the Gap Between Tools for Learning and for Doing Statistics by Amelia Ahlers McNamara Doctor of Philosophy in Statistics University of California, Los Angeles, 2015 Professor Robert L Gould, Co-chair Professor Fredrick R Paik Schoenberg, Co-chair Computers have changed the way we think about data and data analysis. While statistical programming tools have attempted to keep pace with these develop- ments, there is room for improvement in interactivity, randomization methods, and reproducible research. In addition, as in many disciplines, statistical novices tend to have a reduced experience of the true practice. This dissertation discusses the gap between tools for teaching statistics (like TinkerPlots, Fathom, and web applets) and tools for doing statistics (like SAS, SPSS, or R). We posit this gap should not exist, and provide some suggestions for bridging it. Methods for this bridge can be curricular or technological, although this work focuses primarily on the technological. We provide a list of attributes for a modern data analysis tool to support novices through the entire learning-to-doing trajectory. These attributes include easy entry for novice users, data as a first-order persistent object, support for a cycle of exploratory and confirmatory analysis, flexible plot creation, support for randomization throughout, interactivity at every level, inherent visual documenta- tion, simple support for narrative, publishing, and reproducibility, and flexibility ii to build extensions.