Guide to Labeling Your Primary Antibody
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Innova Biosciences A-Z Guide to Fluorochromes
Innova Biosciences A-Z Guide to Fluorochromes Lightning-Link® fluorescent antibody and protein labeling kits allow you to covalently label any antibody in under 20 minutes with only 30 seconds hands-on time. We currently have over 35 labels in our range of fluorescent dyes. This guide provides a general overview of each fluorochrome in our range including their properties, excitation and emission wavelengths and common applications. Allophycocyanin (APC) Atto633 - has a strong absorption at 629nm, high fluorescence at 657nm (extinction coefficient 1.3 Allophycocyanin is a large 105kDa fluorescent x105 cm-1M-1) and high quantum yield. Atto633 can be phycobiliprotein commonly used in FACS analysis. It used as a substitute dye for DY-633, LightCycler Red 640, absorbs at 650nm and emits at 662nm. Alexa Fluor® 633, and Cy 5. Most commonly APC is used for ELISA, flow cytometry, microarrays and other applications that require high Atto700 - has a strong absorption peak at 690nm, with sensitivity but not photostability. an extinction coefficient of 1.2 x105 cm-1M-1, and a maximum emission peak at 725nm. AMCA B-Phycoerythrin (BPE) AMCA (aminomethylcoumarin acetate) is a blue fluorescent dye commonly used in immunofluorescence BPE is a fluorescent protein from the phycobiliprotein microscopy and flow cytometry. It has a maximal family from the cyanbacteria and eukaryotic algae. It has excitation wavelength of 346nm and a maximal emission a strong absorption peak at about 545 nm and has an wavelength of 442nm. emission value of 572 nm. AMCA has good resistance to photobleaching and has B-Phycoerythrin is often thought to be less "sticky" than quite a large Stoke’s shift. -

ZEISS ELYRA Sample Preparation for Superresolution Microscopy – a Quick Guide White Paper
White Paper ZEISS ELYRA Sample Preparation for Superresolution Microscopy – a Quick Guide White Paper ZEISS ELYRA Sample Preparation for Superresolution Microscopy – a Quick Guide Authors: Dr. Sylvia Münter Dr. Yilmaz Niyaz Carl Zeiss Microscopy GmbH, Germany Date: November 2013 General Guidelines for Superresolution Structured Illumination Microscopy (SR-SIM) Sample support In general, we recommend using high-precision coverslips (always no. 1.5 thickness) to mount the samples. High-precision cover glasses feature an exceptionally accurate thickness of 170 ± 5 µm (= 0,170 ± 0,005 mm). Also mind thickness issues when ordering glass-bottom petri dishes or multi-well plates. For cover glasses see: • ZEISS High precision Cover Glasses 18x18 mm; ZEISS order Number: 474030-9010-000 • Marienfeld Superior: www.marienfeld-superior.com • 18x18 mm (Cat.No. 0107032). • 22x22 mm (Cat.No. 0107052). • round with 18 mm diameter (Cat.No. 0117580). • CellPath Ltd UK High performance coverslips no 1.5H 18x18 mm (Cat.No SAN-5018-03A) For glass-bottom petri dishes and multi-well plates see: • Willco (http://www.willcowells.com/) • Lab-tek (offered through Thermo) • MatTek (http://glass-bottom-dishes.com) 2 White Paper Please make sure that the coverslip is centered on the glass slide in order to fit into the sample holder. See below pictures of the ZEISS level adjustable piezo stage insert. Figure 1 ZEISS level adjustable piezo stage insert Fix your cells according to your standard protocol. For SR-SIM imaging, a thoroughly clean glass surface plays a crucial role. Therefore it is beneficial to seal or attach the coverslip in a way that facilitates cleaning with ethanol, without moving the coverslip. -

Dual Color STED Imaging Principles and Tips & Tricks Contents
Nov. 2011 No. 37 CONFOCAL APPLICATION LETTER reSOLUTION Dual Color STED Imaging Principles and Tips & Tricks Contents Stimulated Emission Depletion Microscopy – Basics . 3 Dual Color STED – The Principle . 4 Dye Selection . 6 Leica TCS STED CW . 7 Leica TCS STED . 9 Counterstains . 10 Mounting . 10 Image Acquisition . 11 Image Processing . 13 Conclusions. 15 Cover: HeLa Cells Red: Tubulin, Chromeo 494 Green: Histone H3, ATTO 647N Dual Color STED Imaging Stimulated Emission Depletion Microscopy – Basics Great insights have been obtained from fl uores- (Fig. 1). The fl uorophores within the area of the cence microscopy, but the study of subcellular depletion beam will be forced back to the ground architecture and dynamics can be lost in a blur state. The arrangement of excitation and deple- due to resolution limited by diffraction. Fortu- tion beams achieves an area of emitted fl uores- nately, several approaches to overcome this cence that is smaller than the diffraction limit. limitation have been developed over the last decades1. The resolution of a STED microscope is given by The concept of STimulated Emission Depletion (STED) microscopy, which shrinks the effective focal spot to circumvent the Abbe limit, was fi rst λ described by Stefan Hell in 19942. Currently, the Δx≈ Leica TCS STED and the Leica TCS STED CW em- Ι ploy this technology to provide fast and easy 2NA 1+ access to super-resolution. Ιs where NA is the numerical aperture and λ is the The basic principle is simple: As in confocal STED wavelength. Ι is the intensity of the deple- microscopy, a focal spot is scanned over the tion laser, and Ιs is a dye and the depletion wave- specimen, and the fl uorescence signal emitted length specifi c parameter. -

WO 2016/049544 Al 31 March 2016 (31.03.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/049544 Al 31 March 2016 (31.03.2016) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every G01N 21/64 (2006.01) G01N 21/65 (2006.01) kind of national protection available): AE, AG, AL, AM, G02B 21/00 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (21) Number: International Application DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/US2015/052388 HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (22) International Filing Date: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, 25 September 2015 (25.09.201 5) MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (25) Filing Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (26) Publication Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 62/055,398 25 September 2014 (25.09.2014) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, (71) Applicant: NORTHWESTERN UNIVERSITY TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, [US/US]; 633 Clark Street, Evanston, Illinois 60208 (US). -
WO 2018/055364 Al 29 March 2018 (29.03.2018) W !P O PCT
Thus, a mutant streptavidin subunit conjugated to a fluorescent label via the amino groups will comprise one, two or three fluorescent labels. However, a wild- type streptavidin subunit conjugated to a fluorescent label via the amino groups may comprise more than three fluorescent labels, i.e. four or five fluorescent labels. The term "streptavidin" or "streptavidin protein" as used herein refers to streptavidin comprising four streptavidin subunits. Particularly, streptavidin may be a tetrameric protein comprising four streptavidin subunits where the individual subunits multimerise (e.g. tetramerise) together to form streptavidin. For the tetrameric protein, a nucleic acid sequence may encode an individual subunit(s) which is then translated into a single subunit polypeptide. The individual subunits then multimerise as discussed above to form the tetrameric protein. "Wild-type streptavidin" refers to the wild-type streptavidin protein comprising four wild-type streptavidin subunits as defined further below. Wild-type streptavidin may be in the form of the tetrameric protein formed by the multimerisation of four wild-type streptavidin subunits. Reference to a "streptavidin subunit" or "streptavidin polypeptide" as used herein refers to a single streptavidin subunit which is not multimerised and is not present in tetrameric form. However, a "streptavidin subunit" or "streptavidin polypeptide" as used herein generally refers to a single streptavidin subunit which is capable of multimerisation, e.g. tetramerisation. Thus, the invention further provides a mutant streptavidin protein comprising at least one mutant streptavidin subunit of the invention. The "mutant streptavidin" or "mutant streptavidin protein" of the invention comprises at least one mutant streptavidin subunit of the invention. -
Three-Dimensional Localization of T-Cell Receptors in Relation to Microvilli Using a Combination of Superresolution Microscopies
Three-dimensional localization of T-cell receptors in relation to microvilli using a combination of superresolution microscopies Yunmin Junga, Inbal Rivena, Sara W. Feigelsonb, Elena Kartvelishvilyc, Kazuo Tohyad, Masayuki Miyasakae,f, Ronen Alonb, and Gilad Harana,1 aDepartment of Chemical Physics, Weizmann Institute of Science, Rehovot 76100, Israel; bDepartment of Immunology, Weizmann Institute of Science, Rehovot 76100, Israel; cChemical Research Support, Weizmann Institute of Science, Rehovot 76100, Israel; dDepartment of Anatomy, Kansai University of Health Sciences, Kumatori, Osaka 590-0482, Japan; eInstitute for Academic Initiatives, Osaka University, Suita, Osaka 565-0871, Japan; and fMediCity Research Laboratory, University of Turku, FI-20521, Turku, Finland Edited by Arthur Weiss, University of California, San Francisco, CA, and approved August 8, 2016 (received for review April 3, 2016) Leukocyte microvilli are flexible projections enriched with adhesion recognition event and its consequences, culminating in the forma- molecules. The role of these cellular projections in the ability of T cells to tion of the immunological synapse (IS) (15–17), have been exten- probe antigen-presenting cells has been elusive. In this study, we probe sively studied. Fluorescence imaging studies using a variety of the spatial relation of microvilli and T-cell receptors (TCRs), the major methods, including superresolution microscopy, have demonstrated molecules responsible for antigen recognition on the T-cell membrane. that, even before activation by an antigen, TCRs are not randomly To this end, an effective and robust methodology for mapping distributed on the cell surface, but exist as preformed microclusters membrane protein distribution in relation to the 3D surface structure of an average size of tens of nanometers (18–20). -
Fluoprobes® FT-Fpstd (Standardselectedfluoprobeslabels) ® Fluoprobes Labels
FluoProbes® FT-FPstd_(StandardSelectedFluoProbesLabels) ® FluoProbes Labels Fluoprobes® dyes are designed for labeling biomolecules in advanced RFU fluorescent detection techniques. Applications include multiple labeling, 1,0 FRET, Quenching, polarisation anisotropy fluorescence, and life time resolved fluorescence, with protein as well as with nucleic acids. ® Fluoprobes fluorophores are available as different derivatives for 0,8 conjugation with conventional chemistry methods. This technical sheet presents the main features of selected standard FluoProbes dyes, that are the FP®390 - Em. most popularly used in fluorescence analysis. 0,6 FP®488 - Em. FP®532 - Em. FP®547H - Em. For more information, and information about other FluoProbes labels, FP®565 - Em. FP®594 - Em. please search at www.interchim.com/interchim/customers/ FP®647H - Em. 0,4 FP®682 - Em. or ask FluoProbes at [email protected]. FP®782 - Em. Great FluoProbes labels 0,2 ® Selected standard FluoProbes fluorescent labels for nm 0,0 labeling biomolecules. 350 450 550 650 750 850 £ Product name MW exc\em. max. mol. abs. Comments (key features – see below for more info) cat.number (g·mol-1) (nm) (M-1cm-1) ® FluoProbes 390A Bright blue fluorescence 343.4 390 / 479 24 000 Large stocke's shift A good alternative to AMCA ® FluoProbes 488 Bright green fluorescence, compatible with std filters for FITC/Cy2 Unrivalled stability (photo-stability upon continuous illumination, hence 804 493 / 519 85 000 minimal fading, at PHs, for storage...) Superior alternative to FITC, Cy2, A488 Ideal for confocal microscopy, suits also any other techniques ® FluoProbes 532A Compatible with standard filters for A532 765 532 / 553 115 000 Very bright yellow fluorescence Ideal using the frequency-doubled Nd:YAG laser. -

RESOLFT Nanoscopy with Water-Soluble Synthetic Fluorophores
RESOLFT nanoscopy with water-soluble synthetic fluorophores Dissertation for the award of the degree “Doctor rerum naturalium” of the Georg-August-Universität Göttingen within the doctoral program “Physics of Biological and Complex Systems” of the Georg-August University School of Science (GAUSS) submitted by Philipp Johannes Alt from Trier Göttingen 2017 Members of the Thesis Committee Prof. Dr. Stefan W. Hell (Referee) Department of NanoBiophotonics, Max Planck Institute for Biophysical Chemistry, Göttingen Prof. Dr. Hans-Jürgen Troe (2nd Referee) Institute of Physical Chemistry, Georg-August-Universität Göttingen Dr. Thomas P. Burg Research Group of Biological Micro- and Nanotechnology Max Planck Institute for Biophysical Chemistry, Göttingen Further members of the Examination Board Dr. Melina Schuh Department of Meiosis, Max Planck Institute for Biophysical Chemistry, Göttingen PD Dr. Alexander Egner Department of Optical Nanoscopy, Laser-Laboratorium Göttingen e.V. Prof. Dr. Tim Salditt Research Group for Structure, Dynamics, Assembly and Interaction of Biological Macromolecules, Institute for X-Ray Physics, Georg-August-Universität Göttingen Date of oral examination: 15.12.2017 Abstract Fluorescence microscopy is an important and widely used tool in the life sciences due to its unique ability to observe cellular processes in living specimens with target- specific image contrast. The development of high resolution methods in the far-field has increased its importance further by enabling the visualization of structures fea- turing sizes below the diffraction limit of light. In particular, RESOLFT (reversible saturable optical linear fluorescence transitions) nanoscopy using low light intensities has become a method of choice for live-cell high-resolution fluorescence imaging. RESOLFT nanoscopy requires labels with specialized properties which, to date, have only been observed in reversibly photoswitchable fluorescent proteins (RSFPs). -

Base Spectrale Des Fluorochromes Organiques
Service d'imagerie cellulaire Base spectrale des fluorochromes organiques Avant de choisir un fluorochrome il faut impérativement savoir sur quel(s) instrument(s) sera étudié le signal de fluorescence afin de déterminer quelles en sont les possibilités spectrales en terme d'excitation et d'émission. Avec le microscope confocal SP5-AOBS, vous disposez des raies d'excitation laser : 405, 458, 476, 488, 496, 514, 561 et 633nm. Le système AOBS va permettre d'envoyer la lumière de la longueur d'onde d'excitation choisie vers l'objet et de récupérer toute la fluorescence émise par l'échantillon sur le prisme de diffraction. Le positionnement des fentes de détection devant les différents détecteurs permet de sélectionner à volonté la bande spectrale d'émission dans laquelle on désire réaliser une image. Ainsi le choix des fluorochromes est principalement déterminé par la longueur d'onde d'excitation, et, dans le cas où plusieurs marqueurs sont combinés, par des longueurs d'ondes d'émission suffisamment espacées pour éviter les risques de crosstalk. Les autres critères à prendre en compte sont : - le coefficient d’extinction molaire (on parle aussi d’absorbance molaire) qui est un paramètre d'absorption découlant directement de la loi de Beer- Lambert et qui exprime la variation d'intensité lumineuse (D.O.) en fonction de la concentration molaire de la molécule absorbante et de la longueur du trajet optique. - le rendement quantique f (QE : quantum efficiency ou quantum yield) qui est égal au rapport entre le nombre de photons émis et le nombre de photons reçus dans la zone spectrale d’excitation du fluorochrome. -

Fluoprobes® FT-BB7493
FluoProbes® FT-BB7493 CYanine Carboxyl CYanine fluorophores for labeling biomolecules Introduction A variety of Cyanine dyes has been used to label proteins, nucleic acids and other biomolecules for fluorescence techniques (imaging, biochemical analysis). They replace advantageously the conventional fluorochromes such as Fluorescein(FITC) and rhodamines (TRITC, RRX). CYanine3 can replace orange-fluorescent dyes, like Tetramethylrhodamine (TRITC). Cyanine3 is one of the most broadly used fluorophores which can be detected by various fluorometers, imagers, and microscopes. Due to inherently high extinction coefficient, this dye is also easily detected by naked eye on gels, and in solution. See also alternative superior dye: FluoProbes547H. CYanine3.5 can replace SulfoRhodamine 101. See also alternative superior dye:FluoProbes594. CYanine5 can replace far red red fluorescent dyes. During last years, CYanine5 flurophore has become an incredibly popular label in life science research and diagnostics. Fluorophore emission has maximum in red region, where many CCD detectors have maximum sensitivity, and biological objects have low background. Dye color is very intense, therefore quantity as small as 1 nanomol can be detected in gel electrophoresis by naked eye. See also alternative superior dye: FluoProbes647H CYanine5.5 can replace near infrared fluorescent dyes. See also alternative superior dye: FluoProbes682. CYanine7 is a near infrared red fluorophores used in in vivo imaging applications. See also alternative superior dye: FluoProbes752. CYanine7.5 is a near infrared red fluorophores used for in vivo imaging applications. See also alternative superior dye: FluoProbes800. Sulfo – CYanine dyes are water-soluble form of the CYanine dyes. DiSulfonated forms are the most classic, but some tri- and quadri-sulfonated forms are available as well, for even higher hydrosolubility. -
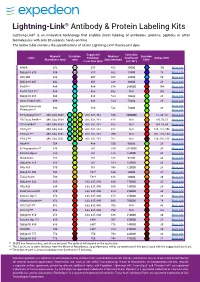
Lightning-Link
Lightning-Link® Antibody & Protein Labeling Kits Lightning-Link® is an innovative technology that enables direct labeling of antibodies, proteins, peptides or other biomolecules with only 30 seconds hands-on time. The below table contains the specifications of all our Lightning-Link® fluorescent dyes. Suggested Extinction Maximal Excitation Maximal Emission Label Excitation Coefficient Stokes Shift Absorbance (nm) color Emission (nm) Color Laser Line (nm) (cm-1M-1) AMCA 352 N/A 355 452 19000 100 More info DyLight® 350 354 N/A 355 432 15000 78 More info Atto 390 388 405 468 24000 80 More info DyLight® 405 402 405 428 30000 26 More info PerCP** 484 488 678 380000 194 More info PerCP/Cy5.5** 484 488 692 N/A 208 More info DyLight® 488 496 488 524 70000 28 More info Alexa Fluor® 488 496 488 524 73000 28 More info Rapid Fluorescein More info 498 488 532 73000 34 Fluorescein** More info R-Phycoerythrin** 498, 544, 566† 488, 532, 561 580 2000000 82, 36, 14 More info PE/Texas Red®** 498, 544, 566† 488, 532, 561 618 N/A 120, 74, 52 More info PE/Atto594** 498, 544, 566† 488, 532, 561 632 N/A 134, 88, 66 More info PE/Cy5** 498, 544, 566† 488, 532, 561 672 N/A 174, 128, 106 More info PE/Cy5.5** 498, 544, 566† 488, 532, 561 700 N/A 202, 156, 134 More info PE/Cy7** 498, 544, 566† 488, 532, 561 782 N/A 284, 238, 216 More info Atto488 504 488 530 90000 26 More info B-Phycoerythrin** 546 561 580 2410000 34 More info Cyanine Dye 3 552 561 576 150000 24 More info Rhodamine 555 561 588 94500 33 More info DyLight® 550 556 561 584 150000 28 More info Atto 565 -

Fluoprobes® FT-FP488 Fluoprobes®488 Labeling Agents
FluoProbes® FT-FP488_ ® FluoProbes 488 labeling agents Product Information A great fluorophore for labeling biomolecules with fluorescent green emission (alternative to FITC) Product name MW exc\em. max. mol. abs. Quantum cat.number (g·mol-1) (nm) (M-1cm-1) yield . M+ (%) Fluoprobes®488 - Carboxyl group 804 590 (L) FP-BA6790, 1mg 493 / 519 80 000 82 Fluoprobes®488 - NHS 981 687 τfl = 3.3 ns (K) FP-BA6800, 1mg ® Fluoprobes 488 - Maleimide CF260(ε260/εmax)= 0.2 1067 712 (M) FP-BA6810, 1mg CF260(ε260/εmax)= 0.1 Fluoprobes®488 - IodoAcetamide A superior alternative to (L) FP-FH9780, 1mg fluoresceins, Cy2, A488... Fluoprobes®488 - Hydrazide Bright green fluorescence (L) FP-B38820, 1mg Fluoprobes®488 - Azide 903 M+: Mass without counter-ion (L) FP-YE4970, 1mg Fluoprobes®488 - Alkyne 740 (L) FP-LV4430, 1mg Fluoprobes®488 Protein Lab.Kit - (V) FP-BE3750, 1 kit Storage: (L): at +4°C (K) : at +4°C (long term at –20°C) (M): at –20°C Fluoprobes®488 is the most popular green emitter fluorescent label, part our the Fluoprobes® dyes series, alternative to FITC, Cy2, A488. Scientific and technical Information - Label FluoProbes®488 is a very bright and photostable green dye* Features: can be excited by any source used for fluoresceins, i.e. the 488 line of Argon laser. compatible with standard filters for Fluoresceins Bright green fluorescence (λexc./λem.: 593/519nm): FP®488 shows elevated extinction coefficient (ε at λmax.: 95 000 M-1cm-1 ) and quantum yield (QY>80%). see comparison [c] Low background compared e.g. with fluoresceins: see comparison [c], and note [b] Absorption and emission spectra Unrivaled photostability at physiological pH-values and upon light exposure (e.g.