The Transuranium Elements: from Neptunium and Plutonium to Element 112
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Table 2.Iii.1. Fissionable Isotopes1
FISSIONABLE ISOTOPES Charles P. Blair Last revised: 2012 “While several isotopes are theoretically fissionable, RANNSAD defines fissionable isotopes as either uranium-233 or 235; plutonium 238, 239, 240, 241, or 242, or Americium-241. See, Ackerman, Asal, Bale, Blair and Rethemeyer, Anatomizing Radiological and Nuclear Non-State Adversaries: Identifying the Adversary, p. 99-101, footnote #10, TABLE 2.III.1. FISSIONABLE ISOTOPES1 Isotope Availability Possible Fission Bare Critical Weapon-types mass2 Uranium-233 MEDIUM: DOE reportedly stores Gun-type or implosion-type 15 kg more than one metric ton of U- 233.3 Uranium-235 HIGH: As of 2007, 1700 metric Gun-type or implosion-type 50 kg tons of HEU existed globally, in both civilian and military stocks.4 Plutonium- HIGH: A separated global stock of Implosion 10 kg 238 plutonium, both civilian and military, of over 500 tons.5 Implosion 10 kg Plutonium- Produced in military and civilian 239 reactor fuels. Typically, reactor Plutonium- grade plutonium (RGP) consists Implosion 40 kg 240 of roughly 60 percent plutonium- Plutonium- 239, 25 percent plutonium-240, Implosion 10-13 kg nine percent plutonium-241, five 241 percent plutonium-242 and one Plutonium- percent plutonium-2386 (these Implosion 89 -100 kg 242 percentages are influenced by how long the fuel is irradiated in the reactor).7 1 This table is drawn, in part, from Charles P. Blair, “Jihadists and Nuclear Weapons,” in Gary A. Ackerman and Jeremy Tamsett, ed., Jihadists and Weapons of Mass Destruction: A Growing Threat (New York: Taylor and Francis, 2009), pp. 196-197. See also, David Albright N 2 “Bare critical mass” refers to the absence of an initiator or a reflector. -

James Chadwick: Ahead of His Time
July 15, 2020 James Chadwick: ahead of his time Gerhard Ecker University of Vienna, Faculty of Physics Boltzmanngasse 5, A-1090 Wien, Austria Abstract James Chadwick is known for his discovery of the neutron. Many of his earlier findings and ideas in the context of weak and strong nuclear forces are much less known. This biographical sketch attempts to highlight the achievements of a scientist who paved the way for contemporary subatomic physics. arXiv:2007.06926v1 [physics.hist-ph] 14 Jul 2020 1 Early years James Chadwick was born on Oct. 20, 1891 in Bollington, Cheshire in the northwest of England, as the eldest son of John Joseph Chadwick and his wife Anne Mary. His father was a cotton spinner while his mother worked as a domestic servant. In 1895 the parents left Bollington to seek a better life in Manchester. James was left behind in the care of his grandparents, a parallel with his famous predecessor Isaac Newton who also grew up with his grandmother. It might be an interesting topic for sociologists of science to find out whether there is a correlation between children educated by their grandmothers and future scientific geniuses. James attended Bollington Cross School. He was very attached to his grandmother, much less to his parents. Nevertheless, he joined his parents in Manchester around 1902 but found it difficult to adjust to the new environment. The family felt they could not afford to send James to Manchester Grammar School although he had been offered a scholarship. Instead, he attended the less prestigious Central Grammar School where the teaching was actually very good, as Chadwick later emphasised. -

An Introduction to Isotopic Calculations John M
An Introduction to Isotopic Calculations John M. Hayes ([email protected]) Woods Hole Oceanographic Institution, Woods Hole, MA 02543, USA, 30 September 2004 Abstract. These notes provide an introduction to: termed isotope effects. As a result of such effects, the • Methods for the expression of isotopic abundances, natural abundances of the stable isotopes of practically • Isotopic mass balances, and all elements involved in low-temperature geochemical • Isotope effects and their consequences in open and (< 200°C) and biological processes are not precisely con- closed systems. stant. Taking carbon as an example, the range of interest is roughly 0.00998 ≤ 13F ≤ 0.01121. Within that range, Notation. Absolute abundances of isotopes are com- differences as small as 0.00001 can provide information monly reported in terms of atom percent. For example, about the source of the carbon and about processes in 13 13 12 13 atom percent C = [ C/( C + C)]100 (1) which the carbon has participated. A closely related term is the fractional abundance The delta notation. Because the interesting isotopic 13 13 fractional abundance of C ≡ F differences between natural samples usually occur at and 13F = 13C/(12C + 13C) (2) beyond the third significant figure of the isotope ratio, it has become conventional to express isotopic abundances These variables deserve attention because they provide using a differential notation. To provide a concrete the only basis for perfectly accurate mass balances. example, it is far easier to say – and to remember – that Isotope ratios are also measures of the absolute abun- the isotope ratios of samples A and B differ by one part dance of isotopes; they are usually arranged so that the per thousand than to say that sample A has 0.3663 %15N more abundant isotope appears in the denominator and sample B has 0.3659 %15N. -

NOBELIUM Element Symbol: No Atomic Number: 102
NOBELIUM Element Symbol: No Atomic Number: 102 An initiative of IYC 2011 brought to you by the RACI KERRY LAMB www.raci.org.au NOBELIUM Element symbol: No Atomic number: 102 The credit for discovering Nobelium was disputed with 3 different research teams claiming the discovery. While the first claim dates back to 1957, it was not until 1992 that the International Union of Pure and Applied Chemistry credited the discovery to a research team from Dubna in Russia for work they did in 1966. The element was named Nobelium in 1957 by the first of its claimed discoverers (the Nobel Institute in Sweden). It was named after Alfred Nobel, a Swedish chemist who invented dynamite, held more than 350 patents and bequeathed his fortune to the establishment of the Nobel Prizes. Nobelium is a synthetic element and does not occur in nature and has no known uses other than in scientific research as only tiny amounts of the element have ever been produced. Nobelium is radioactive and most likely metallic. The appearance and properties of Nobelium are unknown as insufficient amounts of the element have been produced. Nobelium is made by the bombardment of curium (Cm) with carbon nuclei. Its most stable isotope, 259No, has a half-life of 58 minutes and decays to Fermium (255Fm) through alpha decay or to Mendelevium (259Md) through electron capture. Provided by the element sponsor Freehills Patent and Trade Mark Attorneys ARTISTS DESCRIPTION I wanted to depict Alfred Nobel, the namesake of Nobelium, as a resolute young man, wearing the Laurel wreath which is the symbol of victory. -

Th Thorium Compounds with S, Se, Te and B
springer.com Chemistry : Chemistry (general) Brown, David, Wedemeyer, Horst, Buschbeck, Karl-Christian, Keller, Cornelius (Editors-in-chief.) Th Thorium Compounds with S, Se, Te and B The present volume, Thorium C5, deals with the compounds of thorium and sulfur, selenium, tellurium, and boron, as well as with oxoacid compounds of the three chalcogen elements. Thorium borates have already been treated in Thorium C2. In contrast to the corresponding compounds of uranium the thorium sulfides, etc. , do not show any nuclear or other technological application; they are only of academic interest, despite some very interest• ing electronic properties, especially of the 1 : 1 compounds. The thorium-sulfur and the thorium• boron systems in particular were studied in detail, so that we have a clear picture of them, whereas there are still a lot of open questions in the systems Th-Se and Th-Te - not very different from other metal chalcogenide systems. Thorium sulfates are of some technological importance because they are formed in solution during recovery of thorium from monazite by sulfuric acid leaching. The very detailed and critical treatment of the chemical and physical Springer properties of the compounds discussed also enables us to find gaps still remaining in our 8th ed. 1985, XIX, 149 p. knowledge and thus to initiate new research in this field. I want to thank the two authors, Dr. 8th 28 illus., 1 illus. in color. edition Horst Wedemeyer (Karlsruhe) and Dr. David Brown (Harwell), for their excellent contributions, the "Literaturabteilung" of the Karlsruhe Nuclear Research Center for its help in providing reports and other documents difficult to procure, as well as the staff of the Gmelin-Institute, especially to Dr. -

The Transuranium Elements: from Neptunium and Plutonium to Element 112
UCRL-JC- 124728 The Transuranium Elements: From Neptunium and Plutonium to Element 112 Professor Darleane C. Hoffman Lawrence Livermore National Laboratory G. Seaborg Institute for Transactinium Science RECEIVED T. Isotope Sciences Division sEp23 ?9g6 This paper was prepared for submittal to the Conference Proceedings NATO Advanced Study Institute on "Actinides and the Environment" Chania, Crete, Greece July 7-19, 1996 July 26, 1996 DISCWMER This document was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor the University of California nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or the University of California, and shall not be used for advertising or product endorsement purposes. DISCLAIMER Portions of this document may be illegible in electronic image products. Images are produced from the best avaiiable original document. c THE TRANSURANIUM ELEMENTS: FROM NEPTUNIUM AM) PLUTONIUM TO ELEMENT112 DarIeane C. Hoffman Nuclear Science Division & Chemistry Department, University of California, Berkeley, CA 94720 & G. T. Seaborg Institute for ' Transactinium Science, MS-L23 1, LLNL, Livermore, CA 94550. -

Study of Rare Earth Elements, Uranium and Thorium Migration in Rocks from Espinharas Uranium Deposit, Paraiba - Brazil
2009 International Nuclear Atlantic Conference - INAC 2009 Rio de Janeiro,RJ, Brazil, September27 to October 2, 2009 ASSOCIAÇÃO BRASILEIRA DE ENERGIA NUCLEAR - ABEN ISBN: 978-85-99141-03-8 STUDY OF RARE EARTH ELEMENTS, URANIUM AND THORIUM MIGRATION IN ROCKS FROM ESPINHARAS URANIUM DEPOSIT, PARAIBA - BRAZIL Cirilo C.S. Conceição Instituto de Radioproteção e Dosimetria -IRD Av. salvador Allend, S/N - Jacarepaguá Rio de Janeiro, Brazil CEP.: 22780-160 [email protected] ABSTRACT The determination of Rare Earth Elements as natural analogue in patterns geologic has grown as a tool for predicting the long-term safety of nuclear disposal in geological formation. Migration of natural radionuclides is one of the most serious problems in the waste deposit from nuclear fuel cycle. Rare Earth Elements show the same kinetic behavior in rocks as natural radionuclides. This similar property of the analogues allows perform studies and models on the subject of radionuclides migration. The aim of this study was to determine the distribution of Rare Earth Elements in rocks located at Espinharas – Paraíba – Brazil, uranium deposit. In this work are presented the results from the study above the distribution of rare earth elements in function of the degree of mineralized rocks, composition and the conditions of radioactive equilibrium of the uranium and thorium in some fractures on the rocks from radioactive occurrence of Espinharas-Brazil. The results show that there is a correlation of heavy Rare Earth Elements, uranium and Thorium concentrations to oxidation factor of the rocks. However this correlation was not observed for light Rare Earth Elements. It means that heavy Rare earth Elements follow the natural radionuclides in oxidation process of rocks. -

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

Toxicological Profile for Plutonium
PLUTONIUM 1 1. PUBLIC HEALTH STATEMENT This public health statement tells you about plutonium and the effects of exposure to it. The Environmental Protection Agency (EPA) identifies the most serious hazardous waste sites in the nation. These sites are then placed on the National Priorities List (NPL) and are targeted for long-term federal clean-up activities. Plutonium has been found in at least 16 of the 1,689 current or former NPL sites. Although the total number of NPL sites evaluated for this substance is not known, strict regulations make it unlikely that the number of sites at which plutonium is found would increase in the future as more sites are evaluated. This information is important because these sites may be sources of exposure and exposure to this substance may be harmful. When a substance is released from a large area, such as an industrial plant, or from a container, such as a drum or bottle, it enters the environment. This release does not always lead to exposure. You are normally exposed to a substance only when you come in contact with it. You may be exposed by breathing, eating, or drinking the substance, or by skin contact. However, since plutonium is radioactive, you can also be exposed to its radiation if you are near it. External exposure to radiation may occur from natural or man-made sources. Naturally occurring sources of radiation are cosmic radiation from space or radioactive materials in soil or building materials. Man- made sources of radioactive materials are found in consumer products, industrial equipment, atom bomb fallout, and to a smaller extent from hospital waste and nuclear reactors. -

Actinide Ground-State Properties-Theoretical Predictions
Actinide Ground-State Properties Theoretical predictions John M. Wills and Olle Eriksson electron-electron correlations—the electronic energy of the ground state of or nearly fifty years, the actinides interactions among the 5f electrons and solids, molecules, and atoms as a func- defied the efforts of solid-state between them and other electrons—are tional of electron density. The DFT Ftheorists to understand their expected to affect the bonding. prescription has had such a profound properties. These metals are among Low-symmetry crystal structures, impact on basic research in both the most complex of the long-lived relativistic effects, and electron- chemistry and solid-state physics that elements, and in the solid state, they electron correlations are very difficult Walter Kohn, its main inventor, was display some of the most unusual to treat in traditional electronic- one of the recipients of the 1998 behaviors of any series in the periodic structure calculations of metals and, Nobel Prize in Chemistry. table. Very low melting temperatures, until the last decade, were outside the In general, it is not possible to apply large anisotropic thermal-expansion realm of computational ability. And DFT without some approximation. coefficients, very low symmetry crystal yet, it is essential to treat these effects But many man-years of intense research structures, many solid-to-solid phase properly in order to understand the have yielded reliable approximate transitions—the list is daunting. Where physics of the actinides. Electron- expressions for the total energy in does one begin to put together an electron correlations are important in which all terms, except for a single- understanding of these elements? determining the degree to which 5f particle kinetic-energy term, can be In the last 10 years, together with electrons are localized at lattice sites. -
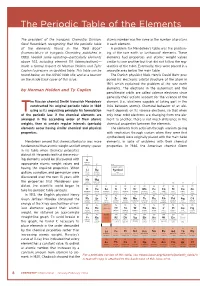
The Periodic Table of the Elements
The Periodic Table of the Elements The president of the Inorganic Chemistry Division, atomic number was the same as the number of protons Gerd Rosenblatt, recognizing that the periodic table in each element. of the elements found in the “Red Book” A problem for Mendeleev’s table was the position- (Nomenclature of Inorganic Chemistry, published in ing of the rare earth or lanthanoid* elements. These 1985) needed some updating—particularly elements elements had properties and atomic weight values above 103, including element 110 (darmstadtium)— similar to one another but that did not follow the reg- made a formal request to Norman Holden and Tyler ularities of the table. Eventually, they were placed in a Coplen to prepare an updated table. This table can be separate area below the main table. found below, on the IUPAC Web site, and as a tear-off The Danish physicist Niels Henrik David Bohr pro- on the inside back cover of this issue. posed his electronic orbital structure of the atom in 1921, which explained the problem of the rare earth by Norman Holden and Ty Coplen elements. The electrons in the outermost and the penultimate orbits are called valence electrons since generally their actions account for the valence of the he Russian chemist Dmitri Ivanovich Mendeleev element (i.e., electrons capable of taking part in the constructed his original periodic table in 1869 links between atoms). Chemical behavior of an ele- Tusing as its organizing principle his formulation ment depends on its valence electrons, so that when of the periodic law: if the chemical elements are only inner orbit electrons are changing from one ele- arranged in the ascending order of their atomic ment to another, there is not much difference in the weights, then at certain regular intervals (periods) chemical properties between the elements. -

Copyrighted Material
ftoc.qrk 5/24/04 1:46 PM Page iii Contents Timeline v de Sitter,Willem 72 Dukas, Helen 74 Introduction 1 E = mc2 76 Eddington, Sir Arthur 79 Absentmindedness 3 Education 82 Anti-Semitism 4 Ehrenfest, Paul 85 Arms Race 8 Einstein, Elsa Löwenthal 88 Atomic Bomb 9 Einstein, Mileva Maric 93 Awards 16 Einstein Field Equations 100 Beauty and Equations 17 Einstein-Podolsky-Rosen Besso, Michele 18 Argument 101 Black Holes 21 Einstein Ring 106 Bohr, Niels Henrik David 25 Einstein Tower 107 Books about Einstein 30 Einsteinium 108 Born, Max 33 Electrodynamics 108 Bose-Einstein Condensate 34 Ether 110 Brain 36 FBI 113 Brownian Motion 39 Freud, Sigmund 116 Career 41 Friedmann, Alexander 117 Causality 44 Germany 119 Childhood 46 God 124 Children 49 Gravitation 126 Clothes 58 Gravitational Waves 128 CommunismCOPYRIGHTED 59 Grossmann, MATERIAL Marcel 129 Correspondence 62 Hair 131 Cosmological Constant 63 Heisenberg, Werner Karl 132 Cosmology 65 Hidden Variables 137 Curie, Marie 68 Hilbert, David 138 Death 70 Hitler, Adolf 141 iii ftoc.qrk 5/24/04 1:46 PM Page iv iv Contents Inventions 142 Poincaré, Henri 220 Israel 144 Popular Works 222 Japan 146 Positivism 223 Jokes about Einstein 148 Princeton 226 Judaism 149 Quantum Mechanics 230 Kaluza-Klein Theory 151 Reference Frames 237 League of Nations 153 Relativity, General Lemaître, Georges 154 Theory of 239 Lenard, Philipp 156 Relativity, Special Lorentz, Hendrik 158 Theory of 247 Mach, Ernst 161 Religion 255 Mathematics 164 Roosevelt, Franklin D. 258 McCarthyism 166 Russell-Einstein Manifesto 260 Michelson-Morley Experiment 167 Schroedinger, Erwin 261 Millikan, Robert 171 Solvay Conferences 265 Miracle Year 174 Space-Time 267 Monroe, Marilyn 179 Spinoza, Baruch (Benedictus) 268 Mysticism 179 Stark, Johannes 270 Myths and Switzerland 272 Misconceptions 181 Thought Experiments 274 Nazism 184 Time Travel 276 Newton, Isaac 188 Twin Paradox 279 Nobel Prize in Physics 190 Uncertainty Principle 280 Olympia Academy 195 Unified Theory 282 Oppenheimer, J.