1440 the Isomeric Hydroxyphenylarsonic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemsherpa 管理対象物質 Ver2.02.00 説明書
chemSHERPA 管理対象物質 Ver2.02.00 説明書 chemSHERPA 事務局 内容 1. はじめに ....................................................................................................................................... 2 2. 管理対象物質リストに関わる用語の定義 ....................................................................................... 3 3. 管理対象物質 Ver2.02.00 について............................................................................................ 4 3.1 管理対象基準 ..................................................................................................................... 4 3.1.1 LR01: 日本 化審法 第一種特定化学物質 ................................................................. 5 3.1.2 LR02︓米国 有害物質規制法(Toxic Substances Control Act︓TSCA)使用禁止 または制限の対象物質(第 6 条) .............................................................................................. 5 3.1.3 LR03︓EU ELV 指令 2000/53/EC ........................................................................... 6 3.1.4 LR04︓EU RoHS 指令 2011/65/EU ANNEX II ..................................................... 6 3.1.5 LR05︓EU POPs 規則 (EU) 2019/1021 ANNEX I ............................................... 7 3.1.6 LR06︓EU REACH 規則 (EC) No 1907/2006 Candidate List of SVHC for Authorisation(認可対象候補物質)および ANNEX XIV(認可対象物質) ........................... 7 3.1.7 LR07︓EU REACH 規則 (EC) No 1907/2006 ANNEX XVII(制限対象物質) . 10 3.1.8 LR08: 欧州 医療機器規則 Annex I 10.4 化学物質 ............................................. 10 3.1.9 IC01︓Global Automotive Declarable Substance List (GADSL) ....................... 11 3.1.10 IC02 ︓ IEC 62474 DB Declarable -

Alfa Laval Black and Grey List, Rev 14.Pdf 2021-02-17 1678 Kb
Alfa Laval Group Black and Grey List M-0710-075E (Revision 14) Black and Grey list – Chemical substances which are subject to restrictions First edition date. 2007-10-29 Revision date 2021-02-10 1. Introduction The Alfa Laval Black and Grey List is divided into three different categories: Banned, Restricted and Substances of Concern. It provides information about restrictions on the use of Chemical substances in Alfa Laval Group’s production processes, materials and parts of our products as well as packaging. Unless stated otherwise, the restrictions on a substance in this list affect the use of the substance in pure form, mixtures and purchased articles. - Banned substances are substances which are prohibited1. - Restricted substances are prohibited in certain applications relevant to the Alfa Laval group. A restricted substance may be used if the application is unmistakably outside the scope of the legislation in question. - Substances of Concern are substances of which the use shall be monitored. This includes substances currently being evaluated for regulations applicable to the Banned or Restricted categories, or substances with legal demands for monitoring. Product owners shall be aware of the risks associated with the continued use of a Substance of Concern. 2. Legislation in the Black and Grey List Alfa Laval Group’s Black and Grey list is based on EU legislations and global agreements. The black and grey list does not correspond to national laws. For more information about chemical regulation please visit: • REACH Candidate list, Substances of Very High Concern (SVHC) • REACH Authorisation list, SVHCs subject to authorization • Protocol on persistent organic pollutants (POPs) o Aarhus protocol o Stockholm convention • Euratom • IMO adopted 2015 GUIDELINES FOR THE DEVELOPMENT OF THE INVENTORY OF HAZARDOUS MATERIALS” (MEPC 269 (68)) • The Hong Kong Convention • Conflict minerals: Dodd-Frank Act 1 Prohibited to use, or put on the market, regardless of application. -

Qingqing Liu
Speciation of arsenic metabolites in chicken meat and in human cells by Qingqing Liu A thesis submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Medical Sciences – Laboratory Medicine and Pathology University of Alberta © Qingqing Liu, 2016 Abstract A phenylarsenical, 3-nitro-4-hydroxyphenylarsonic acid (Roxarsone®, Rox), has been used extensively as a poultry feed additive for over 60 years. However, little is known about the concentrations of different arsenic species present in chicken meat as a consequence of feeding Rox to chickens. The first objective of my thesis research is to study the speciation of Rox in chicken meat, providing relevant information on human exposure to arsenic through the ingestion of chicken meat. The second objective is to study accumulation, transport, and metabolism of Rox and the related arsenic species using human cells, improving a mechanistic understanding. To quantify arsenic species in chicken meat, I first developed an enzyme-assisted extraction method which enhanced the extraction efficiency of arsenic from 28% using water/methanol extraction to 55% using papain digestion. The efficiency was further enhanced to 88% by using ultrasonication combined with the papain digestion. Arsenic species were separated using high performance liquid chromatography (HPLC). Inductively coupled plasma mass spectrometry (ICPMS) and electrospray ionization tandem mass spectrometry (ESI-MS/MS) were used in combination to quantify and identify arsenic species. The method of papain-assisted extraction and HPLC-ICPMS determination achieved detection limits of 1.0-1.8 µg arsenic per kg chicken breast meat for arsenobetaine (AsB), inorganic arsenite (AsIII), dimethylarsinic acid (DMA), monomethylarsonic acid (MMA), inorganic arsenate (AsV), Rox, and N-acetyl-4-hydroxy-m-arsanilic acid (NAHAA). -

And Aminoaryl Arsonic Acids. by Waltera Jacobs, Michablhridelbergbr and Ida P
1580 W. A. JACOSS, &I. .H€:JDEI,SERGER AND IDA P. ROLF. tion. This was especially marked in the use of the barium salt; tests for methyl alcohol and for dimethylamine gave negative results. 2. The solubility of pure helianthine in water is 0.02 g. per liter; the solubilities of all 01 its salts are greater than this. This can indicate that its salts are not completely hydrolyzed. Since solutions of its salts are partially or largely hydrolyzed, as shown in previous studies,' and these colors are not parallel to these solubilities, it may be concluded that the chromoisomerizations of helianthine are largely dependent upon the stabilities of the salts toward mater. 3. KO obvious conclusion can as yet be drawn from studies of the melting points of helianthine salts. The formation of salts by other indicators will be studied. S~ATTLE,WASH [CONTRIBUTION FROM THE LABORATORlES OF THE ROCKEFELLERINSTITUTE FOR MEDICALRESEARCH.] ON NITRO- AND AMINOARYL ARSONIC ACIDS. BY WALTERA JACOBS, MICHABLHRIDELBERGBR AND IDA P. ROLF Received July 9, 1918 In the course of studies on the synthesis of organic arsenic compounds for therapeutic purposes it was found necessary to prepare a number of aminoaryl arsonic acids. It was therefore of importance to find methods which would furnish these substances in amounts sufficient for synthetic work. Of the methods available the application of the Bechamp syn- thesis, first used for the preparation of arsanilic acid, has rendered directly accessible only a limited number, mainly p-aminoaryl arsonic acids. Tn order to obtain a- and m-amino arsonic acids it has usually been necessary to employ indirect methods. -

Factory Farm Nation 2015 EDITION About Food & Water Watch
Factory Farm Nation 2015 EDITION About Food & Water Watch ood & Water Watch FKDPSLRQVKHDOWK\IRRGDQGFOHDQZDWHUIRUDOO:HVWDQGXSWRFRUSRUDWLRQVWKDWSXWSURȴWV Fbefore people, and advocate for a democracy that improves people’s lives and protects our environment. )RRG :DWHU:DWFKKDVVWDWHDQGUHJLRQDORɝFHVDFURVVWKHFRXQWU\WRKHOSHQJDJHFRQFHUQHGFLWL]HQVRQWKH LVVXHVWKH\FDUHDERXW)RUWKHPRVWXSWRGDWHFRQWDFWLQIRUPDWLRQIRURXUȴHOGRɝFHVYLVLWfoodandwaterwatch.org. Los Angeles, California Oakland, California Colorado Florida 3000 S. Robertson Boulevard 1814 Franklin Street 1740 High Street 1044 NE 15th Avenue Suite 255 Suite 1100 Denver, CO 80218 Fort Lauderdale, FL 33304 Los Angeles, CA 90034 Oakland, CA 94612 (720) 449-7505 (954) 372-1881 (323) 843-8450 (510) 922-0720 Illinois Iowa Maine Maryland 811 W. Evergreen Avenue 505 Fifth Avenue 533 Congress Street 3121 St. Paul Street Suite 401 Suite 818 Portland, ME 04101 Suite 28 Chicago, IL 60642 Des Moines, IA 50309 (207) 619-5845 Baltimore, MD 21218 (773) 796-6088 (515) 344-4834 (410) 394-7650 Michigan New Jersey New Mexico New York 2727 Second Avenue 100 Bayard Street 7804 Pan American 68 Jay Street Suite 136 Suite 202 East Freeway NE #2 Suite 713 Detroit, MI 48201-2654 New Brunswick, NJ 08901 Albuquerque, NM 87109 Brooklyn, NY 11201 (313) 486-1356 (732) 839-0860 (505) 633-7366 (718) 943-9085 North Carolina Ohio Oregon Pennsylvania 801 Gilbert Street 103 William H. Taft Road 917 SW Oak Street 1501 Cherry Street Suite 204 Cincinnati, OH 45219 Suite 404 Second Floor Durham, NC 27701 (513) 394-6257 Portland, OR 97205 Philadelphia, PA 19102 (919) 794-6380 (971) 266-4528 (267) 428-1903 1DWLRQDO2ɝFH 1616 P Street, NW Suite 300 Washington, DC 20036 (202) 683-2500 Copyright © May 2015 by Food & Water Watch. -

Estimation of Total Arsenic in Maize, Fish and Human Urine from Some Parts of Amansie West District in the Ashanti Region of Gh
ESTIMATION OF TOTAL ARSENIC IN MAIZE, FISH AND HUMAN URINE FROM SOME PARTS OF AMANSIE WEST DISTRICT IN THE ASHANTI REGION OF GHANA By NUWORDZRO, JOANITTA A Thesis submitted to the Department of Chemistry, College of Science, Kwame Nkrumah University of Science and Technology, Kumasi in partial fulfillment of the requirements for the award of degree MASTER OF PHILOSOPHY (Environmental Chemistry) JUNE, 2013 DECLARATION I hereby declare that this thesis is my own research work and that, to the best of my knowledge, it contains no material previously published by another person or material which has been accepted for the award of any degree of the University, except where due acknowledgement has been made in the text. …………………………… …………………………… Joanitta Nuwordzro Date (Student) I declare that I supervised this student in undertaking this study; I hereby confirm that the student had my permission to present this thesis for assessment. …………………………… …………………………… Dr. Sylvestern K. Twumasi Date (Supervisor) …………………………… …………………………… Dr. Ray B. Voegborlo Date (Head of Department) ii DEDICATION This is dedicated to my best friend Isaac Annor who has supported me in every way in my academic pursuit and also to my mother Miss Monica Agbley and brother P. Y. Boateng who have been there for me. iii ACKNOWLEDGEMENTS Glory to God, the Father, the Son and the Holy Spirit. Great things He has done. I will always thank you Oh Lord for your grace and mercy. My special thanks go to Dr. S.K Twumasi, my supervisor for his support, fatherly love and excellent supervision. My sincerest appreciation goes to Mr. Nash Bentil of Ghana Atomic Energy Commission who led me in the use of the hydride generation atomic absorption spectrophotometerfor this work, Miss Ernestina Boapong of Keegan Resources Ghana Limited for her help during the sampling of fish along the river Offin and also Dr G. -

Velux Green Supply Chain Management for the Protection of the Environment and Human Health and Sustainable Development
VELUX RESTRICTED SUBSTANCE MANAGEMENT STANDARD VELUX GREEN SUPPLY CHAIN MANAGEMENT FOR THE PROTECTION OF THE ENVIRONMENT AND HUMAN HEALTH AND SUSTAINABLE DEVELOPMENT. VELUX RESTRICTED SUBSTANCES MANAGEMENT STANDARD (VRSMS) 1 September 2020 _____________________________________________________________________________________ INTRODUCTION For more than 75 years the VELUX Group (hereafter “VELUX”) has created better living environments by bringing daylight and fresh air into people's homes and commercial buildings all over the world. Our products help create bright, healthy, energy-efficient places in which to live, work, learn and play. VELUX has manufacturing and sales operations in more than 40 countries, with an extensive distribution network. Our products include a wide range of residential applications such as roof windows, skylights, flat roof windows and a wide range of commercial applications such as modular skylights with a wide range of installation solutions, roller shutters, blinds, decorative elements and remote controls. VELUX designs our products to minimise their impact on the environment and human health during sourcing, manufacture, use and disposal. To ensure that, we – as a downstream user - take a proactive and precautious approach to managing restricted substances and substances of high concern, VELUX has established a global chemical management process. Further, VELUX has established a management process for sustainable development. Our suppliers must be committed to support VRSMS generally and must comply with all environmental laws and regulations applicable for their business and must comply with VELUX internal restrictions in addition to the restrictions required by law or regulation (“VELUX Thresholds”) for the protection of the environment and human health as well as sustainable development. Further our suppliers must support us in our compliance with all environmental laws and regulations applicable for our business, including the issuance of compliance certificates and notification of first supply of substances. -

Atom Trapping Vapor Generation Atomic Absorption Spectrometry for Inorganicarsenic Speciation Analysis a Thesis Submitted To
ATOM TRAPPING VAPOR GENERATION ATOMIC ABSORPTION SPECTROMETRY FOR INORGANICARSENIC SPECIATION ANALYSIS A THESIS SUBMITTED TO THE GRADUATE SCHOOL OF NATURAL AND APPLIED SCIENCES OF MIDDLE EAST TECHNICAL UNIVERSITY BY NJAW NJIE IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN CHEMISTRY AUGUST 2014 i Approval of the thesis: ATOM TRAPPING VAPOR GENERATION ATOMIC ABSORPTION SPECTROMETRY FOR INORGANIC ARSENIC SPECIATION ANALYSIS Submitted by NJAW NJIE in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry Department, Middle East Technical University by, Prof. Dr. Canan Özgen _____________________ Dean, Graduate School of Natural and Applied Sciences Prof. Dr. İlker Özkan _____________________ Head of Department, Chemistry Prof. Dr. O. Yavuz Ataman _____________________ Supervisor, Chemistry Dept., METU Examining Committee Members: Assoc. Prof. G. Inci Gokmen _____________________ Chemistry Dept., METU Prof. Dr. O. Yavuz Ataman ____________________ Supervisor, Chemistry Dept., METU Prof. Dr. Mürvet Volkan _____________________ Chemistry Dept., METU Prof. Dr. Orhan Acar _____________________ Analitik Kimya Anabilim Dali Eczacilik Fak., Gazi Uni. Prof. Dr. Nusret Ertaş _____________________ Analitik Kimya Anabilim Dali Eczacilik Fak., Gazi Uni. Date: __________________ ii I hereby declare that all information in this document has been obtained and presented in accordance with academic rules and ethical conduct. I also declare that, as required by these rules and conduct, I have fully cited and referenced all material and results that are not original to this work. Name, Last name: Njaw Njie Signature: iv ABSTRACT ATOM TRAPPING VAPOR GENERATION ATOMIC ABSORPTION SPECTROMETRY FOR INORGANICARSENIC SPECIATION ANALYSIS Njie, Njaw Ph.D., Department of Chemistry Supervisor: Prof. Dr. -

Correspondence
CORE Metadata, citation and similar papers at core.ac.uk Provided by Research Commons@Waikato 1 Correspondence to: Professor B K Nicholson Chemistry Department University of Waikato Private Bag 3105 Hamilton New Zealand Email: [email protected] Fax: 64 7 838 4219 Substituted phenylarsonic acids; structures and spectroscopy. Nicholas C Lloyd, Hugh W. Morgan, Brian K Nicholson* and Ron S. Ronimus School of Science and Engineering, University of Waikato, Private Bag 3105, Hamilton 3240, New Zealand. _____________________________________________________________________ Synopsis Spectroscopic data for fifteen substituted phenylarsonic acids are reported, together with the X-ray structures of five examples which form extensive H-bonded networks in the crystals. Pictogram N O O O H H H N H N+ O H O H - O As O H H O As O H H O As N _____________________________________________________________________ 2 Abstract. Full NMR and ESI-MS spectra, and differential scanning calorimeter data are presented for fifteen substituted phenylarsonic acids, including two new fluoro- substituted examples. X-ray crystal structure determinations of five examples (phenylarsonic acid and the 4-fluoro-, 4-fluoro-3-nitro-, 3-amino-4-hydroxy- and 3- amino-4-methoxy- substituted derivatives) were determined and the H-bonding crystal-packing patterns analysed. 1. Introduction. As part of a project to re-investigate the composition and biological activity of the historically-important arsenical remedy for syphilis, Salvarsan, we required a series of substituted arylarsonic acids, RAsO3H2 [1]. There is revived interest in organo-arsenic compounds with the recognition that they are still of pharmaceutical use [2] despite their obvious toxicity, and with a developing understanding of the biological pathways for activity [3]. -
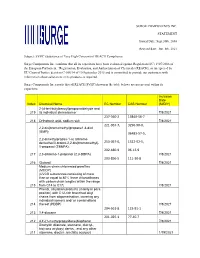
SURGE COMPONENTS INC. STATEMENT Issued Date: Sept.30Th, 2016 Revised Date: Jun. 8Th, 2021 Subject: SVHC (Substance of Very High
SURGE COMPONENTS INC. STATEMENT Issued Date: Sept.30th, 2016 Revised date: Jun. 8th, 2021 Subject: SVHC (Substance of Very High Concern) of REACH Compliance Surge Components Inc. confirms that all its capacitors have been evaluated against Regulation (EC) 1907/2006 of the European Parliament, “Registration, Evaluation, and Authorization of Chemicals (REACH), as interpreted by EU Court of Justice decision C-106/14 of 10 September 2015 and is committed to provide our customers with information about substances in its products as required. Surge Components Inc.reports that all REACH SVHC(shown in the table below) are not present within its capacitors. Inclusion Date Index Chemical Name EC Number CAS Number (M/D/Y) 2-(4-tert-butylbenzyl)propionaldehyde and 219 its individual stereoisomer 7/8/2021 237-560-2 13840-56-7 218 Orthoboric acid, sodium salt 7/8/2021 221-967-7, 3296-90-0, 2,2-bis(bromomethyl)propane1,3-diol (BMP); 36483-57-5, 2,2-dimethylpropan-1-ol, tribromo derivative/3-bromo-2,2-bis(bromomethyl)- 253-057-0, 1522-92-5, 1-propanol (TBNPA); 202-480-9 96-13-9 217 2,3-dibromo-1-propanol (2,3-DBPA) 7/8/2021 203-856-5 111-30-8 216 Glutaral 7/8/2021 Medium-chain chlorinated paraffins (MCCP) (UVCB substances consisting of more than or equal to 80% linear chloroalkanes with carbon chain lengths within the range 215 from C14 to C17) 7/8/2021 Phenol, alkylation products (mainly in para position) with C12-rich branched alkyl chains from oligomerisation, covering any individual isomers and/ or combinations 214 thereof (PDDP) 7/8/2021 204-661-8 123-91-1 213 1,4-dioxane 7/8/2021 201-025-1 77-40-7 212 4,4'-(1-methylpropylidene)bisphenol 7/8/2021 Dioctyltin dilaurate, stannane, dioctyl-, bis(coco acyloxy) derivs., and any other 211 stannane, dioctyl-, bis(fatty acyloxy) 1/19/2021 derivs. -

THE FATE and SPECIATION of ARSENIC in SOILS and POULTRY PRODUCTION SYSTEMS by Jennifer M. Seiter a Dissertation Submitted To
THE FATE AND SPECIATION OF ARSENIC IN SOILS AND POULTRY PRODUCTION SYSTEMS by Jennifer M. Seiter A dissertation submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Plant and Soil Sciences Spring 2009 Copyright 2009 Jennifer Seiter All Rights Reserved UMI Number: 3360255 INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleed-through, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. ______________________________________________________________ UMI Microform 3360255 Copyright 2009 by ProQuest LLC All rights reserved. This microform edition is protected against unauthorized copying under Title 17, United States Code. _______________________________________________________________ ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, MI 48106-1346 THE FATE AND SPECIATION OF ARSENIC IN SOILS AND POULTRY PRODUCTION SYSTEMS by Jennifer M. Seiter Approved: ___________________________________________________________ Blake C. Meyers, Ph.D. Chairperson of the Department of Plant and Soil Sciences Approved: ___________________________________________________________ Robin W. Morgan, Ph.D. Dean of the College of Agriculture and Natural Resources Approved: ___________________________________________________________ Debra H. Norris, M.S. Vice Provost for Graduate and Professional Education ii I certify that I have read this dissertation and that in my opinion it meets the academic and professional standard required by the University as a dissertation for the degree of Doctor of Philosophy. -

Arsenic and Arsenic Compounds
United States Office of Air Quality EPA-454/R-98-013 Environmental Protection Planning And Standards June 1998 Agency Research Triangle Park, NC 27711 AIR EPA LOCATING AND ESTIMATING AIR EMISSIONS FROM SOURCES OF ARSENIC AND ARSENIC COMPOUNDS L & E This report has been reviewed by the Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency, and has been approved for publication. Mention of trade names and commercial products does not constitute endorsement or recommendation for use. EPA-454/R-98-013 ii TABLE OF CONTENTS Section Page LIST OF TABLES........................................................... viii LIST OF FIGURES........................................................... xi EXECUTIVE SUMMARY.................................................... xiv 1.0 PURPOSE OF DOCUMENT ............................................ 1-1 2.0 OVERVIEW OF DOCUMENT CONTENTS................................ 2-1 3.0 BACKGROUND ...................................................... 3-1 3.1 Physical And Chemical Nature Of Arsenic And Arsenic Compounds ....... 3-1 3.1.1 Inorganic Compounds ...................................... 3-3 3.1.2 Organic Arsenic Compounds ................................ 3-10 3.2 Fate Of Arsenic ................................................ 3-14 3.2.1 Fate of Arsenic in Soil ..................................... 3-14 3.2.2 Fate of Arsenic in Water ................................... 3-15 3.2.3 Fate of Arsenic in Air ...................................... 3-16 3.2.4 Fate of Arsenic in Plants and Food