Solving the Phase Problem in Protein Electron Crystallography: Multiple Isomorphous Replacement and Anomalous Dispersion As Alternatives to Imaging
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Introduction to Phasing Crystallography ISSN 0907-4449
research papers Acta Crystallographica Section D Biological Introduction to phasing Crystallography ISSN 0907-4449 Garry L. Taylor When collecting X-ray diffraction data from a crystal, we Received 30 August 2009 measure the intensities of the diffracted waves scattered from Accepted 22 February 2010 a series of planes that we can imagine slicing through the Centre for Biomolecular Sciences, University of St Andrews, St Andrews, Fife KY16 9ST, crystal in all directions. From these intensities we derive the Scotland amplitudes of the scattered waves, but in the experiment we lose the phase information; that is, how we offset these waves when we add them together to reconstruct an image of our Correspondence e-mail: [email protected] molecule. This is generally known as the ‘phase problem’. We can only derive the phases from some knowledge of the molecular structure. In small-molecule crystallography, some basic assumptions about atomicity give rise to relationships between the amplitudes from which phase information can be extracted. In protein crystallography, these ab initio methods can only be used in the rare cases in which there are data to at least 1.2 A˚ resolution. For the majority of cases in protein crystallography phases are derived either by using the atomic coordinates of a structurally similar protein (molecular replacement) or by finding the positions of heavy atoms that are intrinsic to the protein or that have been added (methods such as MIR, MIRAS, SIR, SIRAS, MAD, SAD or com- binations of these). The pioneering work of Perutz, Kendrew, Blow, Crick and others developed the methods of isomor- phous replacement: adding electron-dense atoms to the protein without disturbing the protein structure. -

System Design and Verification of the Precession Electron Diffraction Technique
NORTHWESTERN UNIVERSITY System Design and Verification of the Precession Electron Diffraction Technique A DISSERTATION SUBMITTED TO THE GRADUATE SCHOOL IN PARTIAL FULFILLMENT OF THE REQUIREMENTS for the degree DOCTOR OF PHILOSOPHY Field of Materials Science and Engineering By Christopher Su-Yan Own EVANSTON, ILLINOIS First published on the WWW 01, August 2005 Build 05.12.07. PDF available for download at: http://www.numis.northwestern.edu/Research/Current/precession.shtml c Copyright by Christopher Su-Yan Own 2005 All Rights Reserved ii ABSTRACT System Design and Verification of the Precession Electron Diffraction Technique Christopher Su-Yan Own Bulk structural crystallography is generally a two-part process wherein a rough starting structure model is first derived, then later refined to give an accurate model of the structure. The critical step is the deter- mination of the initial model. As materials problems decrease in length scale, the electron microscope has proven to be a versatile and effective tool for studying many problems. However, study of complex bulk structures by electron diffraction has been hindered by the problem of dynamical diffraction. This phenomenon makes bulk electron diffraction very sensitive to specimen thickness, and expensive equip- ment such as aberration-corrected scanning transmission microscopes or elaborate methodology such as high resolution imaging combined with diffraction and simulation are often required to generate good starting structures. The precession electron diffraction technique (PED), which has the ability to significantly reduce dynamical effects in diffraction patterns, has shown promise as being a “philosopher’s stone” for bulk electron diffraction. However, a comprehensive understanding of its abilities and limitations is necessary before it can be put into widespread use as a standalone technique. -

Optical Ptychographic Phase Tomography
University College London Final year project Optical Ptychographic Phase Tomography Supervisors: Author: Prof. Ian Robinson Qiaoen Luo Dr. Fucai Zhang March 20, 2013 Abstract The possibility of combining ptychographic iterative phase retrieval and computerised tomography using optical waves was investigated in this report. The theoretical background and historic developments of ptychographic phase retrieval was reviewed in the first part of the report. A simple review of the principles behind computerised tomography was given with 2D and 3D simulations in the following chapters. The sample used in the experiment is a glass tube with its outer wall glued with glass microspheres. The tube has a diameter of approx- imately 1 mm and the microspheres have a diameter of 30 µm. The experiment demonstrated the successful recovery of features of the sam- ple with limited resolution. The results could be improved in future attempts. In addition, phase unwrapping techniques were compared and evaluated in the report. This technique could retrieve the three dimensional refractive index distribution of an optical component (ideally a cylindrical object) such as an opitcal fibre. As it is relatively an inexpensive and readily available set-up compared to X-ray phase tomography, the technique can have a promising future for application at large scale. Contents List of Figures i 1 Introduction 1 2 Theory 3 2.1 Phase Retrieval . .3 2.1.1 Phase Problem . .3 2.1.2 The Importance of Phase . .5 2.1.3 Phase Retrieval Iterative Algorithms . .7 2.2 Ptychography . .9 2.2.1 Ptychography Principle . .9 2.2.2 Ptychographic Iterative Engine . -

Electron Crystallography of Ultrathin 3D Protein Crystals: Atomic Model with Charges
Electron crystallography of ultrathin 3D protein crystals: Atomic model with charges Koji Yonekura (米倉 功治)a,b, Kazuyuki Kato (加藤 一幸)c, Mitsuo Ogasawara (小笠原 光雄)b, Masahiro Tomita (富田 正弘)b,d, and Chikashi Toyoshima (豊島 近)b,1 aBiostructural Mechanism Laboratory, RIKEN SPring-8 Center, 1-1-1 Kouto, Sayo, Hyogo 679-5148, Japan; bInstitute of Molecular and Cellular Biosciences, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo, 113-0032, Japan; cHitachi High-Tech Fielding Corporation, 4-28-8 Yotsuya, Shinjuku-ku, Tokyo, 160-0004, Japan; and dHitachi High-Technologies Corporation, 1-24-14 Nishi-Shinbashi, Minato-ku, Tokyo, 105-8717, Japan Contributed by Chikashi Toyoshima, January 23, 2015 (sent for review August 28, 2014) Membrane proteins and macromolecular complexes often yield F and G). These features of Coulomb potential maps result from crystals too small or too thin for even the modern synchrotron the fact that atomic scattering factors for electrons vary consid- X-ray beam. Electron crystallography could provide a powerful erably over a range of spatial frequency depending on the means for structure determination with such undersized crystals, charged state (Fig. 1A) and can become close to zero or even − as protein atoms diffract electrons four to five orders of magni- negative (e.g., for O , Fig. 1A). An advantageous consequence is tude more strongly than they do X-rays. Furthermore, as electron that it is possible to determine experimentally the charged states crystallography yields Coulomb potential maps rather than elec- of protein residues and metals. As proteins use metals of different tron density maps, it could provide a unique method to visualize ionic states for many purposes, notably for catalysis and electron the charged states of amino acid residues and metals. -

X-Ray Crystallography
X-ray Crystallography Prof. Leonardo Scapozza Pharmceutical Biochemistry School of Pharmaceutical Sciences University of Geneva, University of Lausanne E-mail: [email protected] Aim • Introduce the students to X-ray crystallography • Give the students the tools to “evaluate” a X-ray structure based scientific paper 1 Outline • The History of X-ray • The Principle of X-ray • The Steps towards the 3D structure – Crystallization – X-ray diffraction and data collection – From Pattern of Diffraction to Electron Density – X-ray structure quality assessment An extract of a structure paper 2.1. Crystallization • The hTK1 was cloned as N-terminal thrombin-cleavable His6–tagged fusion protein missing 14 amino acids of the N-terminus and 40 amino acids of the C–terminus of the wild type hTK1 sequence of 234 amino acids (this construct is further on called hTK1). The purified hTK1, consisting of residues 15-194 of the wild type sequence plus an N–terminal extension of 15 residues containing a His6–tag, was eluted from gel filtration column at a concentration of approximate 7 mg/ml with a buffer containing 5 mM Tris at pH 7.2, 10 mM NaCl and 10 mM DTT. For protein crystallization we used the hanging drop method at 23°C. Initial conditions for crystallization were found using Crystal screen Cryo no. 40 (Hampton Research). The protein solution was mixed in a 1:1 ratio with crystallization buffer (0.095 mM tri–sodium citrate pH 5.5, 12% PEG 4000, 10% isopropanol) to set up drops of 6 μl. The reservoir contained 500 μl of crystallization buffer. -

Recent Advances in Electron Crystallography
pISSN 2287-5123·eISSN 2287-4445 https://doi.org/10.9729/AM.2017.47.3.160 Review Article Recent Advances in Electron Crystallography Jeong Min Chung†, Sangmin Lee†, Hyun Suk Jung* Department of Biochemistry, College of Natural Sciences, Kangwon National University, Chuncheon 24341, Korea Electron crystallography has been used as the one of powerful tool for studying the structure of biological macromolecules at high resolution which is sufficient to provide †These authors contributed equally details of intramolecular and intermolecular interactions at near-atomic level. Previously to this work. it commonly uses two-dimensional crystals that are periodic arrangement of biological molecules, however recent studies reported a novel technical approach to electron *Correspondence to: crystallography of three-dimensional crystals, called micro electron-diffraction (MicroED) Jung HS, which involves placing the irregular and small sized protein crystals in a transmission Tel: +82-33-250-8513 electron microscope to determine the atomic structure. In here, we review the advances in Fax: +82-33-259-9363 electron crystallography techniques with several recent studies. Furthermore, we discuss E-mail: [email protected] the future direction of this structural approach. Received August 7, 2017 Revised September 6, 2017 Key Words: Electron crystallography, Protein structure, Transmission electron microscopy, Accepted September 8, 2017 Micro-electron diffraction, Structural biology INTRODUCTION crystals found during the screening process (Bill et al., 2011). Since early 1940s, electron diffraction has been used to solve The ultimate goal of structural biology is to understand the crystallographic problems (Bendersky & Gayle, 2001). the protein function and its physiological mechanisms by The basic principle of electron crystallography is similar determining the three-dimensional (3D) structure. -

Electron Crystallography of Aquaporins
Portland State University PDXScholar Chemistry Faculty Publications and Presentations Chemistry 7-2008 Electron Crystallography of Aquaporins Simeon Andrews University of Washington Tacoma Steve Reichow [email protected] Tamir Gonen Howard Hughes Medical Institute Follow this and additional works at: https://pdxscholar.library.pdx.edu/chem_fac Part of the Biochemistry, Biophysics, and Structural Biology Commons, and the Chemistry Commons Let us know how access to this document benefits ou.y Citation Details Andrews, S., Reichow, S. L., & Gonen, T. (2008). Electron crystallography of aquaporins. IUBMB life, 60(7), 430-436. This Post-Print is brought to you for free and open access. It has been accepted for inclusion in Chemistry Faculty Publications and Presentations by an authorized administrator of PDXScholar. Please contact us if we can make this document more accessible: [email protected]. NIH Public Access Author Manuscript IUBMB Life. Author manuscript; available in PMC 2009 June 4. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: IUBMB Life. 2008 July ; 60(7): 430±436. doi:10.1002/iub.53. Electron Crystallography of Aquaporins Simeon Andrews, Steve L. Reichow, and Tamir Gonen Department of Biochemistry, University of Washington, Seattle, WA, USA Summary Aquaporins are a family of ubiquitous membrane proteins that form a pore for the permeation of water. Both electron and X-ray crystallography played major roles in determining the atomic structures of a number of aquaporins. This review focuses on electron crystallography, and its contribution to the field of aquaporin biology. We briefly discuss electron crystallography and the two-dimensional crystallization process. -

Electron Crystallography: Imaging and Single-Crystal Diffraction from Powders
feature articles Acta Crystallographica Section A Foundations of Electron crystallography: imaging and single-crystal Crystallography diffraction from powders ISSN 0108-7673 Xiaodong Zoua,b* and Sven Hovmo¨llera Received 28 September 2007 Accepted 16 November 2007 aStructural Chemistry, Stockholm University, SE-106 91 Stockholm, Sweden, and bBerzelii Centre EXSELENT on Porous Materials, Stockholm University, SE-106 91 Stockholm, Sweden. Correspondence e-mail: [email protected] The study of crystals at atomic level by electrons – electron crystallography – is an important complement to X-ray crystallography. There are two main advantages of structure determinations by electron crystallography compared to X-ray diffraction: (i) crystals millions of times smaller than those needed for X-ray diffraction can be studied and (ii) the phases of the crystallographic structure factors, which are lost in X-ray diffraction, are present in transmission- electron-microscopy (TEM) images. In this paper, some recent developments of electron crystallography and its applications, mainly on inorganic crystals, are shown. Crystal structures can be solved to atomic resolution in two dimensions as well as in three dimensions from both TEM images and electron diffraction. Different techniques developed for electron crystallography, including three- dimensional reconstruction, the electron precession technique and ultrafast electron crystallography, are reviewed. Examples of electron-crystallography # 2008 International Union of Crystallography applications are given. There is in principle no limitation to the complexity of Printed in Singapore – all rights reserved the structures that can be solved by electron crystallography. 1. Introduction Henderson, 1975) using Fourier-transform-based image processing to get both the crystallographic structure-factor Electron diffraction (ED) of crystals was discovered in 1927, amplitudes and phases from the TEM images and retrieve the only 15 years after the discovery of X-ray diffraction. -

Pulsed EPR Determination of Water Accessibility to Spin-Labeled Amino Acid Residues in Lhciib
1124 Biophysical Journal Volume 96 February 2009 1124–1141 Pulsed EPR Determination of Water Accessibility to Spin-Labeled Amino Acid Residues in LHCIIb A. Volkov,† C. Dockter,‡ T. Bund,‡ H. Paulsen,‡ and G. Jeschke§* †Max-Planck Institute for Polymer Research, Mainz, Germany; ‡Institute of General Botany, Johannes Gutenberg University, Mainz, Germany; and §Laboratory of Physical Chemistry, Swiss Federal Institute of Technology, Zu¨rich, Switzerland ABSTRACT Membrane proteins reside in a structured environment in which some of their residues are accessible to water, some are in contact with alkyl chains of lipid molecules, and some are buried in the protein. Water accessibility of residues may change during folding or function-related structural dynamics. Several techniques based on the combination of pulsed elec- tron paramagnetic resonance (EPR) with site-directed spin labeling can be used to quantify such water accessibility. Accessibility parameters for different residues in major plant light-harvesting complex IIb are determined by electron spin echo envelope modulation spectroscopy in the presence of deuterated water, deuterium contrast in transversal relaxation rates, analysis of longitudinal relaxation rates, and line shape analysis of electron-spin-echo-detected EPR spectra as well as by the conventional techniques of measuring the maximum hyperfine splitting and progressive saturation in continuous-wave EPR. Systematic comparison of these parameters allows for a more detailed characterization of the environment of the spin-labeled residues. These techniques are applicable independently of protein size and require ~10–20 nmol of singly spin-labeled protein per sample. For a residue close to the N-terminus, in a domain unresolved in the existing x-ray structures of light-harvesting complex IIb, all methods indicate high water accessibility. -
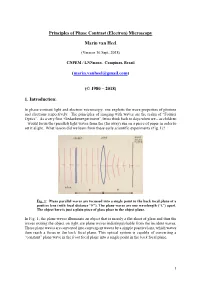
Principles of the Phase Contrast (Electron) Microscopy
Principles of Phase Contrast (Electron) Microscopy Marin van Heel (Version 16 Sept. 2018) CNPEM / LNNnano, Campinas, Brazil ([email protected]) (© 1980 – 2018) 1. Introduction: In phase-contrast light and electron microscopy, one exploits the wave properties of photons and electrons respectively. The principles of imaging with waves are the realm of “Fourier Optics”. As a very first “Gedankenexperiment”, let us think back to days when we – as children – would focus the (parallel) light waves from the (far away) sun on a piece of paper in order to set it alight. What lesson did we learn from these early scientific experiments (Fig. 1)? Fig. 1: Plane parallel waves are focussed into a single point in the back focal plane of a positive lens (with focal distance “F”). The plane waves are one wavelength (“λ”) apart. The object here is just a plain piece of glass place in the object plane. In Fig. 1, the plane waves illuminate an object that is merely a flat sheet of glass and thus the waves exiting the object on right are plane waves indistinguishable from the incident waves. These plane waves are converted into convergent waves by a simple positive lens, which waves then reach a focus in the back focal plane. This optical system is capable of converting a “constant” plane wave in the front focal plane into a single point in the back focal plane. 1 Fig. 2: A single point scatterer in the object plane leads to secondary (“scattered”) concentric waves emerging from that point in the object. Since the object is placed in the front focal plane, these scattered or “diffracted” secondary waves become plane waves hitting the back focal plane of the system. -
Phase Problem in X-Ray Crystallography, and Its Solution Reciprocal Directions and Spacings from the Real Lattice
Phase Problem in X-ray Secondary article Crystallography, and Its Article Contents . The Phase Problem in X-ray Crystallography Solution . Patterson Methods . Direct Methods Kevin Cowtan, University of York, UK . Multiple Isomorphous Replacement . Multiwavelength Anomalous Dispersion (MAD) X-ray crystallography can provide detailed information about the structure of biological . Molecular Replacement molecules if the ‘phase problem’ can be solved for the molecule under study. The . Phase Improvement phase problem arises because it is only possible to measure the amplitude of diffraction . Summary spots: information on the phase of the diffracted radiation is missing. Techniques are available to reconstruct this information. The Phase Problem in X-ray called the unit cell. It is convenient to define crystal axes a, b Crystallography and c defining the unit cell in three dimensions. Scattering along directions reflecting the lattice repeat will be X-ray diffraction provides one of the most important tools reinforced by every repeat of the unit cell, and will be for examining the three-dimensional (3D) structure of strong; scattering along all other directions will be weak. biological macromolecules. The physics of diffraction As a result, the full diffraction pattern of the crystal is a requires that in order to resolve features of atomic pattern of spots, forming a three-dimensional lattice with structure it is necessary to employ radiation with a wavelength of the order of atomic spacing or smaller. X- rays have suitable wavelength, and interact with the Scattered waves are electrons of the atoms. However, the interaction between out of phase X-rays and electrons is weak, and such energetic radiation causes ionization of the constituent atoms of the molecule, damaging the molecule under study. -

Three-Dimensional Electron Crystallography of Protein Microcrystals Dan Shi†, Brent L Nannenga†, Matthew G Iadanza†, Tamir Gonen*
RESEARCH ARTICLE elife.elifesciences.org Three-dimensional electron crystallography of protein microcrystals Dan Shi†, Brent L Nannenga†, Matthew G Iadanza†, Tamir Gonen* Janelia Farm Research Campus, Howard Hughes Medical Institute, Ashburn, United States Abstract We demonstrate that it is feasible to determine high-resolution protein structures by electron crystallography of three-dimensional crystals in an electron cryo-microscope (CryoEM). Lysozyme microcrystals were frozen on an electron microscopy grid, and electron diffraction data collected to 1.7 Å resolution. We developed a data collection protocol to collect a full-tilt series in electron diffraction to atomic resolution. A single tilt series contains up to 90 individual diffraction patterns collected from a single crystal with tilt angle increment of 0.1–1° and a total accumulated electron dose less than 10 electrons per angstrom squared. We indexed the data from three crystals and used them for structure determination of lysozyme by molecular replacement followed by crystallographic refinement to 2.9 Å resolution. This proof of principle paves the way for the implementation of a new technique, which we name ‘MicroED’, that may have wide applicability in structural biology. DOI: 10.7554/eLife.01345.001 Introduction X-ray crystallography depends on large and well-ordered crystals for diffraction studies. Crystals are *For correspondence: gonent@ solids composed of repeated structural motifs in a three-dimensional lattice (hereafter called ‘3D janelia.hhmi.org crystals’). The periodic structure of the crystalline solid acts as a diffraction grating to scatter the †These authors contributed X-rays. For every elastic scattering event that contributes to a diffraction pattern there are ∼10 inelastic equally to this work events that cause beam damage (Henderson, 1995).