Design and Practical Usage of Web Biological Databases for the Annotation and Classification of Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Diverse Deep-Sea Anglerfishes Share a Genetically Reduced Luminous
RESEARCH ARTICLE Diverse deep-sea anglerfishes share a genetically reduced luminous symbiont that is acquired from the environment Lydia J Baker1*, Lindsay L Freed2, Cole G Easson2,3, Jose V Lopez2, Dante´ Fenolio4, Tracey T Sutton2, Spencer V Nyholm5, Tory A Hendry1* 1Department of Microbiology, Cornell University, New York, United States; 2Halmos College of Natural Sciences and Oceanography, Nova Southeastern University, Fort Lauderdale, United States; 3Department of Biology, Middle Tennessee State University, Murfreesboro, United States; 4Center for Conservation and Research, San Antonio Zoo, San Antonio, United States; 5Department of Molecular and Cell Biology, University of Connecticut, Storrs, United States Abstract Deep-sea anglerfishes are relatively abundant and diverse, but their luminescent bacterial symbionts remain enigmatic. The genomes of two symbiont species have qualities common to vertically transmitted, host-dependent bacteria. However, a number of traits suggest that these symbionts may be environmentally acquired. To determine how anglerfish symbionts are transmitted, we analyzed bacteria-host codivergence across six diverse anglerfish genera. Most of the anglerfish species surveyed shared a common species of symbiont. Only one other symbiont species was found, which had a specific relationship with one anglerfish species, Cryptopsaras couesii. Host and symbiont phylogenies lacked congruence, and there was no statistical support for codivergence broadly. We also recovered symbiont-specific gene sequences from water collected near hosts, suggesting environmental persistence of symbionts. Based on these results we conclude that diverse anglerfishes share symbionts that are acquired from the environment, and *For correspondence: that these bacteria have undergone extreme genome reduction although they are not vertically [email protected] (LJB); transmitted. -

Chapter 3 Polyphasic Taxonomy: Idiomarina and Pseudoidiomarina
PhD Thesis 2011 Chapter 3 Polyphasic taxonomy: Idiomarina and Pseudoidiomarina species Jay Siddharth Page 82 PhD Thesis 2011 Chapter 3 Polyphasic taxonomy: Idiomarina and Pseudoidiomarina species. Chapter synopsis. An attempt to cultivate the extremophiles from Lonar lake was made using standard microbiological approaches. This employed an array of media combinations for the initial screening process. This was followed by 16S rRNA based identification of the isolates. The isolates that had very low similarity was subjected to polyphasic taxonomic investigation. This resulted in the isolation of a new species whose proposed names are Idiomarina lonarii and Pseudoidiomarina lonarii. Jay Siddharth Page 83 PhD Thesis 2011 Introduction: Genus Idiomarina Two strains, KMM 227T and KMM 231T, were isolated from seawater sampled at a depths of 4000 and 5000 m, respectively, in the northwestern part of the Pacific Ocean. The new isolates were similar in their phenotypic properties to other marine aerobic proteobacteria, including Alteromonas, Pseudoalteromonas and Marinomonas, but they were characterized by several salient differences from known genera: morphological peculiarities, inability to use carbohydrates as the sole source of carbon and energy, and unusual cellular fatty acid composition. On the basis of phenotypic, chemotaxonomic and phylogenetic data, these marine deep-sea bacteria were classified as Idiomarina abyssalis and Idiomarina zobellii (177). Phylogeny Comparative 16S rDNA sequence analysis of Idiomarina strains KMM 227 and KMM -

Pseudidiomarina Taiwanensis Gen. Nov., Sp. Nov., a Marine Bacterium
International Journal of Systematic and Evolutionary Microbiology (2006), 56, 899–905 DOI 10.1099/ijs.0.64048-0 Pseudidiomarina taiwanensis gen. nov., sp. nov., a marine bacterium isolated from shallow coastal water of An-Ping Harbour, Taiwan, and emended description of the family Idiomarinaceae Wen Dar Jean,1 Wung Yang Shieh2 and Hsiu-Hui Chiu2 Correspondence 1Center for General Education, Leader University, No. 188, Sec. 5, An-Chung Rd, Tainan, Wung Yang Shieh Taiwan [email protected] 2Institute of Oceanography, National Taiwan University, PO Box 23-13, Taipei, Taiwan Two strains of heterotrophic, aerobic, marine bacteria, designated strains PIT1T and PIT2, were isolated from sea-water samples collected at the shallow coastal region of An-Ping Harbour, Tainan, Taiwan. Both strains were Gram-negative. Cells grown in broth cultures were straight rods that were non-motile, lacking flagella. Both strains required NaCl for growth and exhibited optimal growth at 30–35 6C, 1–4 % NaCl and pH 8. They grew aerobically and were incapable of anaerobic growth by fermentation of glucose or other carbohydrates. Cellular fatty acids were predominantly iso-branched, with C15 : 0 iso and C17 : 0 iso representing the most abundant components. The DNA G+C contents of strains PIT1T and PIT2 were 49?3 and 48?6 mol%, respectively. Phylogeny based on 16S rRNA gene sequences, together with data from phenotypic and chemotaxonomic characterization, revealed that the two isolates could be assigned to a novel genus in the family Idiomarinaceae, for which the name Pseudidiomarina gen. nov. is proposed. Pseudidiomarina taiwanensis sp. nov. is the type species of the novel genus (type strain PIT1T=BCRC 17465T=JCM 13360T). -

Isolation of Halophilic Bacteria from Inland Petroleum- Producing Wells
100 • FINE FOCUS, VOL. 3 (2) HALOPHILIC BACTERIA • 101 ISOLATION OF HALOPHILIC BACTERIA FROM INLAND PETROLEUM- PRODUCING WELLS MAEDGEN Q. LINDSEY & JENNIFER R. HUDDLESTON* BIOLOGY DEPARTMENT, ABILENE CHRISTIAN UNIVERSITY, ABILENE, TX 79699 MANUSCRIPT RECEIVED 28 MARCH 2017; ACCEPTED 20 MAY 2017 Copyright 2017, Fine Focus. All rights reserved. 102 • FINE FOCUS, VOL. 3 (2) HALOPHILIC BACTERIA • 103 a Pennsylvanian era channel-sand deposition organic osmotic solutes to remain isotonic located in Central Northern Texas (5), and thus relative to the environment (24). slightly older and deeper than the Flippen ABSTRACT Limestone, measuring at a depth of 2000 feet Due to the range of salinities, temperatures, at the oil well location. Saltwater produced at and possible carbon sources, oil felds and The goals of this study were to isolate microorganisms these sites is pumped to the surface with oil their related infrastructure pose a potential from oil well-produced water, identify the and gas, where the components are separated treasure trove of microbial diversity. microorganisms, and test the microorganisms’ salt and eventually utilized or discarded. Previously identifed microorganisms isolated tolerance. Saltwater collected from two well locations from oil well associated environments producing from different zones in Jones County, Texas, A consequence of the prevalence of oil include Marinobacter aquaeolei, which was was spread onto Mannitol Salt Agar (MSA). Isolates drilling is hydrocarbon contamination of described after being isolated from the head showed a 16S rDNA gene sequence identity of 99% soils and groundwater. Microbial-mediated of an offshore oil rig off the coast of Vung with Idiomarina baltica and Marinobacter persicus. -

Pseudidiomarina Marina Sp. Nov. and Pseudidiomarina Tainanensis Sp
International Journal of Systematic and Evolutionary Microbiology (2009), 59, 53–59 DOI 10.1099/ijs.0.001180-0 Pseudidiomarina marina sp. nov. and Pseudidiomarina tainanensis sp. nov. and reclassification of Idiomarina homiensis and Idiomarina salinarum as Pseudidiomarina homiensis comb. nov. and Pseudidiomarina salinarum comb. nov., respectively Wen Dar Jean,1 Tsung-Yen Leu,2 Chung-Yi Lee,2 Ta-Jen Chu,3 Silk Yu Lin4 and Wung Yang Shieh4 Correspondence 1Center for General Education, Leader University, No. 188, Sec. 5, An-Chung Rd, Tainan, Taiwan, Wung Yang Shieh ROC [email protected] 2Department of Microbiology, Soochow University, Taipei, Taiwan, ROC 3Department of Leisure and Recreational Management, Chung Hua University, 30, Tung-Hsiang, Hsinchu, Taiwan, ROC 4Institute of Oceanography, National Taiwan University, PO Box 23-13, Taipei, Taiwan, ROC Two Gram-negative strains of heterotrophic, aerobic, marine bacteria, designated PIM1T and PIN1T, were isolated from seawater samples collected from the shallow coastal region of An-Ping Harbour, Tainan, Taiwan. Cells grown in broth cultures were straight rods and non-motile. The two isolates required NaCl for growth and grew optimally at 30–35 6C and 2–5 % NaCl. They grew aerobically and were not capable of anaerobic growth by fermentation of glucose or other carbohydrates. The cellular fatty acids were predominantly iso-branched, with iso-C15 : 0 (17.0– 21.4 %), iso-C17 : 0 (18.2–21.0 %) and iso-C17 : 1v9c (15.7–16.6 %) as the most abundant components. The predominant isoprenoid quinone was Q-8 (95.2–97.1 %). Strains PIM1T and PIN1T had DNA G+C contents of 46.6 and 46.9 mol%, respectively. -
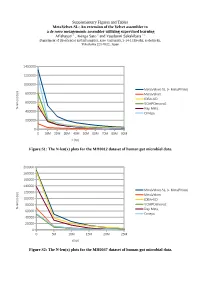
Supplementary Figures and Tables Metavelvet-SL
Supplementary Figures and Tables MetaVelvet-SL: An extension of the Velvet assembler to a de novo metagenomic assembler utilizing supervised learning Afiahayati 1 , Kengo Sato 1 and Yasubumi Sakakibara 1∗ Department of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan 1400000 1200000 1000000 MetaVelvet-SL (+ MetaPhlAn) ) p 800000 b MetaVelvet ( ) x ( IDBA-UD n 600000 e SOAPDenovo2 l - N Ray Meta 400000 Omega 200000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S1: The N-len(x) plots for the MH0012 dataset of human gut microbial data. 200000 180000 160000 140000 MetaVelvet-SL (+ MetaPhlAn) ) 120000 p b MetaVelvet ( ) 100000 x IDBA-UD ( n e l 80000 SOAPDenovo2 - N Ray Meta 60000 Omega 40000 20000 0 0 5M 10M 15M 20M 25M x(bp) Figure S2: The N-len(x) plots for the MH0047 dataset of human gut microbial data. 600000 500000 400000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 300000 x IDBA-UD ( n e l SOAPDenovo2 - N 200000 Ray Meta Omega 100000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S3: The N-len(x) plots for the SRS017227 dataset of human gut microbial data. 800000 700000 600000 500000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 400000 x IDBA-UD ( n e l SOAPDenovo2 - 300000 N Ray Meta 200000 Omega 100000 0 0 5M 10M 15M 20M 25M 30M 35M 40M 45M x (bp) Figure S4: The N-len(x) plots for the SRS018661 dataset of human gut microbial data. Formula to identify unique nodes in Velvet (Zerbino and Birney, 2008) ̄x2 ρ2− log2 2 F ( ̄x ,n,ρ )= +n 2 2 where ̄x = the coverage of node ρ = expected coverage of subgraph n = the length of node A node is “unique”, if its F > 5 . -

Aliidiomarina Taiwanensis Gen. Nov., Sp. Nov., a Marine Bacterium Isolated from Shallow Coastal Water from Bitou Harbour, Taiwan
Aliidiomarina taiwanensis gen. nov., sp. nov., a marine bacterium isolated from shallow coastal water from Bitou Harbour, Taiwan Ssu-Po Huang1, Hsiao-Yun Chang1, Jwo-Sheng Chen2, Wen Dar Jean3 and Wung Yang Shieh1* (1) Institute of Oceanography, National Taiwan University, PO Box 23-13, Taipei, Taiwan (2) College of Health Care, China Medical University, No. 91, Shyue-Shyh Rd, Taichung, Taiwan (3) Center for General Education, Leader University, No. 188, Sec. 5, An-Chung Rd, Tainan, Taiwan *Wung Yang Shieh (Corresponding author) Email: [email protected]; Tel.: +886 2 33661398; Fax: +886 2 23626092. Abstract A Gram-negative, heterotrophic, aerobic, marine bacterium, designated strain AIT1T, was isolated from a seawater sample collected in the shallow coastal region of Bitou Harbour, Taipei County, Taiwan. Cells grown in broth cultures were straight to slightly curved rods that were motile by means of a single polar flagellum. Strain AIT1T required NaCl for growth and grew optimally at 30-40 °C and 1.5-5% NaCl. It grew aerobically and was incapable of anaerobic growth by fermentation of glucose or other carbohydrates. Isoprenoid quinones consisted of Q-8 (95.2 %) and Q-9 (4.8 %). Cellular fatty acids were predominantly iso-branched, including iso-C17:0 (26.5 %), iso-C17:19c (25.9 %) and iso-C15:0 (20.5 %). The DNA G + C content was 51.5 mol%. A phylogenetic analysis based on 16S rRNA gene sequences showed that strain AIT1T formed a distinct lineage within the class Gammaproteobacteria and exhibited highest levels of sequence similarity with species of Idiomarina in the family Idiomarinaceae (91.5-93.9 %). -

Supplementary Materials
Supplementary Materials Supporting methods Sequence comparison to DNA isolation kit blank and drilling fluid (For Costa Rica sediment samples) Because DNA concentrations were very low in many of the sediment samples, and PCR tests indicated that in a few of the samples, if present at all, DNA may not be in high enough amounts to overcome the “background” DNA from the DNA extraction kits, a representative DNA extraction kit blank was sequenced along with all other samples. To remove any signal from the extraction kit in all samples, as well as to remove any samples whose genuine DNA was not in high enough abundance to overcome the extraction kit background, sequence results from the SILVA pipeline were processed initially as follows: 1. Classification of reads was examined at the “fully expanded” taxonomic depth from the SILVA pipeline output, and all lineages present in the extraction blank in any amount were flagged. 2. To account for sequencing error in classification, further lineages were added to the flagged ones by going up in taxonomic level to “order” and flagging every sequence identified as being from the same order as any sequence present in the extraction blank. There were a few cases where the taxonomy of sequences in the extraction blank did not go down as far as the level of "order", and for those, the most specific level identified above order was used to assess any further matches. For example, if the sequence was classified down to "class," then any remaining sequences in that class would also be removed. Those cases -

Idiomarina Piscisalsi Sp. Nov., from Fermented Fish (Pla-Ra) in Thailand
J. Gen. Appl. Microbiol., 59, 385‒391 (2013) Short Communication Idiomarina piscisalsi sp. nov., from fermented fish pla-ra( ) in Thailand Jaruwan Sitdhipol,1,† Wonnop Visessanguan,2 Soottawat Benjakul,3 Pattaraporn Yukphan,4 and Somboon Tanasupawat1,* 1 Department of Biochemistry and Microbiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok 10330, Thailand 2 Food Biotechnology Laboratory, National Center for Genetic Engineering and Biotechnology (BIOTEC), Pathumthani 12120, Thailand 3 Department of Food Technology, Faculty of Agro-Industry, Prince of Songkla University, Hat Yai, Songkhla 90112, Thailand 4 BIOTEC Culture Collection (BCC), National Center for Genetic Engineering and Biotechnology (BIOTEC), Pathumthani 12120, Thailand (Received August 17, 2012; Accepted June 18, 2013) Key Words—fermented fish; Idiomarina piscisalsi; γ-Proteobacteria; 16S rRNA gene The family Idiomarinaceae, class Gammaproteobac- Korea (Choi and Cho, 2005), Idiomarina homiensis teria, was proposed by Ivanova et al. (2004) according from seashore sand in Korea (Kwon et al., 2006), Idi- to the phylogenetic relationships of marine Alteromon- omarina salinarum from a marine solar saltern in Korea as-like bacteria. The family formerly contained two (Yoon et al., 2007), Idiomarina insulisalsae sp. nov., genera, Idiomarina and Pseudidiomarina, which were isolated from the soil of a sea salt evaporation pond, proposed by Ivanova et al. (2000) and Jean et al. Idiomarina marina, Idiomarina maritima and Idiomarina (2006), respectively. At the -

Ca–Mg Kutnahorite and Struvite Production by Idiomarina Strains at Modern Seawater Salinities
Available online at www.sciencedirect.com Chemosphere 72 (2008) 465–472 www.elsevier.com/locate/chemosphere Ca–Mg kutnahorite and struvite production by Idiomarina strains at modern seawater salinities Marı´a Teresa Gonza´lez-Mun˜oz a,*, Concepcio´n De Linares b, Francisca Martı´nez-Ruiz c, Fernando Morcillo a, Daniel Martı´n-Ramos d, Jose´ Marı´a Arias a a Departamento de Microbiologı´a, Facultad de Ciencias, Campus Fuentenueva, Universidad de Granada, 18002 Granada, Spain b Departamento de Bota´nica, Facultad de Ciencias, Campus Fuentenueva, Universidad de Granada, 18002 Granada, Spain c Instituto Andaluz de Ciencias de la Tierra (CSIC-UGR), Facultad de Ciencias, Campus Fuentenueva, Universidad de Granada, 18002 Granada, Spain d Departamento de Mineralogı´a y Petrologı´a. Facultad de Ciencias, Campus Fuentenueva, Universidad de Granada, 18002 Granada, Spain Received 25 October 2007; received in revised form 4 February 2008; accepted 6 February 2008 Available online 19 March 2008 Abstract The production of Mg-rich carbonates by Idiomarina bacteria at modern seawater salinities has been investigated. With this objective, four strains: Idiomarina abyssalis (strain ATCC BAA-312), Idiomarina baltica (strain DSM 15154), Idiomarina loihiensis (strains DSM 15497 and MAH1) were used. The strain I. loihiensis MAH1 is a new isolate, identified in the scope of this work. The four moderately halophilic strains precipitated struvite (NH4MgPO4 Á 6H2O) crystals that appear encased by small Ca–Mg kutnahorite [CaMg(CO3)2] spheres and dumbbells, which are also regularly distributed in the bacterial colonies. The proportion of Ca–Mg kutnahorite produced by the bacteria assayed ranged from 50% to 20%, and I. -

Idiomarina Fontislapidosi Sp. Nov. and Idiomarina Ramblicola Sp. Nov., Isolated from Inland Hypersaline Habitats in Spain
International Journal of Systematic and Evolutionary Microbiology (2004), 54, 1793–1797 DOI 10.1099/ijs.0.63172-0 Idiomarina fontislapidosi sp. nov. and Idiomarina ramblicola sp. nov., isolated from inland hypersaline habitats in Spain M. Jose´ Martı´nez-Ca´novas, Victoria Be´jar, Fernando Martı´nez-Checa, Rafael Pa´ez and Emilia Quesada Correspondence Microbial Exopolysaccharide Research Group, Department of Microbiology, Faculty of Emilia Quesada Pharmacy, Campus Universitario de Cartuja, University of Granada, 18071 Granada, Spain [email protected] Two bacterial strains, F23T and R22T, have been isolated from hypersaline habitats in Ma´laga (S. Spain) and Murcia (E. Spain). The novel strains, similar to previously described Idiomarina species, are slightly curved rods, Gram-negative, chemo-organotrophic, strictly aerobic and motile by a single polar flagellum. Both strains produce catalase and oxidase. They hydrolyse aesculin, gelatin, casein, Tween 20, Tween 80 and DNA but not starch or tyrosine. The strains differ from the hitherto described Idiomarina species in their capacity to produce extracellular polysaccharides and their different patterns of carbon sources and antimicrobial susceptibility. They are moderate halophiles capable of growing in NaCl concentrations of 0?5 to 25 % w/v, the optimum being 3–5 % w/v. Cellular fatty acids are predominantly iso-branched. The main fatty acids in strain FP23T are 15 : 0 iso (26?75 %), 16 : 1v7c (11?33 %) and 16 : 0 (11?73 %) whilst 15 : 0 iso (24?69 %), 17 : 0 iso (12?92 %) and 17 : 1v9c (11?03 %) are predominant in strain R22T. The DNA G+C composition is 46?0 mol% in strain FP23T and 48?7 mol% in strain R22T.