103 ORGAN1 C ANALYSIS. Estimation of Acetone in Smokeless
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemistry (55)
156 Chemistry (55) Introduction 3) To expose the students to various emerging According to NCF 2005, the new and new areas of chemistry and apprise them updated curriculum is introduced at +2 stage. with their relevance in their future studies There is a need to provide the sufficient and their applications in various spheres conceptual background of chemistry which will of chemical sciences and technology. help the students to appear for different common 4) To equip students to face various changes entrance test at the state level and the national related to health, nutrition, environment, level. This new syllabus will make them population, weather, industries and competent to meet the challenges of academic agriculture. and professional courses like medicine, 5) To develop problem solving skills in engineering, technology, etc, after the +2 stage. students. The syllabus is comparable to the international 6) To expose the students to different level. processes used in industries and their The syllabus contains areas like physical, technological applications. organic, inorganic, industrial, analytical and 7) To apprise students with interface of polymer chemistry. The upgraded syllabus has chemistry with other disciplines of science taken care of new formulations and such as physics, biology, geology, nomenclature of elements, compounds and engineering, etc. IUPAC units of physical quantities. New nomenclature, symbols and formulations, Std. XI (Theory) fundamental concepts, modern techniques are given importance. Unit 1: Some Basic Concepts of Chemistry Objectives : General Introduction: Importance and The broad objectives of teaching scope of chemistry. Historical approach to Chemistry at Higher Secondary stage are to particulate nature of matter, laws of help the learners : chemical combination, Dalton’s atomic 1) To promote understanding of basic facts theory : concept of elements, atoms and and concepts in chemistry while retaining molecules. -

Download Download
Test for Acetone in Urine 189 AN IMPROVED TEST FOR ACETONE IN URINE. R. E. Lyons and J. T. Brundage, Indiana University. Lieben's test for acetone 1 depends upon the formation of iodoform when potassium iodide, iodine solution, and a few drops of sodium hydroxide solution are added to an acetone containing mixture. The iodoform is recognized by its distinctive odor and, microscopically, by the 1 star, or hexagonal crystals. The test is not specific since both ethyl alcohol and acetic aldehyde also react with these reagents to yield iodo- form. This sometimes leads to erroneous results because of alcohol formed through sugar fermentation in diabetic urine. The difficulty is obviated" by substituting ammonium hydroxide for the caustic alkali as proposed by Gunning'5 in a modification of the Lieben Test. In either test the reaction is much more sensitive if a urine dis- tillate is used. The distillation not only frees the acetone from non- volatile interfering substances, but converts some acetonacetic (di-acetic) 4 acid, if present, into acetone. Protein interferes and, if present, the separation of acetone by distillation or aeration is necessary. M. KohlthofF' states that 100 mg. each of potassium iodide and chlor- amine T, 10-20 drops of 4N ammonium hydroxide and 10 cc. of a solu- tion of one part acetone in 10,000 parts of 2 per cent ethyl alcohol when warmed to 60 °C. gave an iodoform precipitate in two hours. The object of our investigation has been to determine (a) whether this reaction could be applied as a specific test for acetone in urine, (b) what urinary constituents or preservatives interfere, (c) if the necessity of distillation, or aeration, of the urine could be dispensed with, and (d) the conditions for attaining the maximum sensitiveness of the reaction. -

Laboratory 23: Properties of Aldehydes and Ketones
Laboratory 23: Properties of Aldehydes and Ketones Introduction Aldehydes and Ketones represent an important class of organic molecules containing a carbonyl carbon. In this experiment you will study the chemical properties of aldehydes and ketones. Solubility in water, and organic solvents, combustibility, and reactivity with various chemical reagents will be examined. Discussion Structure of Aldehydes and Ketones Aldehydes and Ketones are organic compounds containing a carbonyl carbon (R−C−O−R') (Figure 1 below) functional group. Carboxylic Acids and Esters also contain a carbonyl carbon, and will be explored in a future experiment. The carbonyl carbon is a polar group with the carbon having a slight excess of positive charge and the oxygen atom having a slight excess of negative charge. Chemical Properties Aldehydes and ketones are created by the mild oxidation of primary and secondary alcohols. One such method to oxidize alcohols is with copper (II) oxide. Upon heading, copper wire (Cu0) in an open flame leads to the formation of copper (II) oxide. The copper (II) oxide is then reacted with an alcohol to form an aldehyde or ketone, copper (I) oxide and water. [O] Primary Alcohol −−! Aldehyde [O] Secondary Alcohol −−! Ketone Chemically aldehydes and ketones both contain a carbonyl carbon and thus have similar chemical reactivities. However, aldehydes are more susceptible to oxidation because of the hydrogen atom attached to the carbonyl group. This is the basis for distinguishing between these two classes of compounds. Several tests are useful for differentiating between aldehydes and ketones. The first test is referred to as the Tollens' or Silver Mirror test. -

Pharmaceutical Services Division and the Clinical Research Centre Ministry of Health Malaysia
A publication of the PHARMACEUTICAL SERVICES DIVISION AND THE CLINICAL RESEARCH CENTRE MINISTRY OF HEALTH MALAYSIA MALAYSIAN STATISTICS ON MEDICINES 2008 Edited by: Lian L.M., Kamarudin A., Siti Fauziah A., Nik Nor Aklima N.O., Norazida A.R. With contributions from: Hafizh A.A., Lim J.Y., Hoo L.P., Faridah Aryani M.Y., Sheamini S., Rosliza L., Fatimah A.R., Nour Hanah O., Rosaida M.S., Muhammad Radzi A.H., Raman M., Tee H.P., Ooi B.P., Shamsiah S., Tan H.P.M., Jayaram M., Masni M., Sri Wahyu T., Muhammad Yazid J., Norafidah I., Nurkhodrulnada M.L., Letchumanan G.R.R., Mastura I., Yong S.L., Mohamed Noor R., Daphne G., Kamarudin A., Chang K.M., Goh A.S., Sinari S., Bee P.C., Lim Y.S., Wong S.P., Chang K.M., Goh A.S., Sinari S., Bee P.C., Lim Y.S., Wong S.P., Omar I., Zoriah A., Fong Y.Y.A., Nusaibah A.R., Feisul Idzwan M., Ghazali A.K., Hooi L.S., Khoo E.M., Sunita B., Nurul Suhaida B.,Wan Azman W.A., Liew H.B., Kong S.H., Haarathi C., Nirmala J., Sim K.H., Azura M.A., Asmah J., Chan L.C., Choon S.E., Chang S.Y., Roshidah B., Ravindran J., Nik Mohd Nasri N.I., Ghazali I., Wan Abu Bakar Y., Wan Hamilton W.H., Ravichandran J., Zaridah S., Wan Zahanim W.Y., Kannappan P., Intan Shafina S., Tan A.L., Rohan Malek J., Selvalingam S., Lei C.M.C., Ching S.L., Zanariah H., Lim P.C., Hong Y.H.J., Tan T.B.A., Sim L.H.B, Long K.N., Sameerah S.A.R., Lai M.L.J., Rahela A.K., Azura D., Ibtisam M.N., Voon F.K., Nor Saleha I.T., Tajunisah M.E., Wan Nazuha W.R., Wong H.S., Rosnawati Y., Ong S.G., Syazzana D., Puteri Juanita Z., Mohd. -

Safety Data Sheet Iodoform Compound Paint
SAFETY DATA SHEET IODOFORM COMPOUND PAINT 1. IDENTIFICATION OF THE SUBSTANCE/PREPARATION AND THE COM PANY: PRODUCT NAME: IODOFORM COMPOUND PAINT PART No.: M028 SUPPLIER: J M Loveridge plc Southbrook Road, Southampton Hampshire SO15 1BH Tel: 023 8022 2008 Fax: 023 8022 2117 2. COMPOSITION/INFORMATION ON INGREDIENTS: NAME CONTENT CAS No.: EINECS Nr.: CLASSIFICATION DIETHYL ETHER 60-100 % 60-29-7 200-467-2 Xn ,Fx R-12, 19, 22, 66, 67 IODOFORM 10-30 % 75-47-8 200-874-5 Xn R-20/21/22, 36 The Full Text for all R-Phrases are Displayed in Section 16 3. HAZARDS IDENTIFICATION: Extremely flammable. Harmful if swallowed. Repeated exposure may cause skin dryness or cracking. Vapours may cause drowsiness and dizziness. 4. FIRST AID MEASURES: GENERAL: IN ALL CASES OF DOUBT OR WHEN SYMPTOMS PERSIST, ALWAYS SEEK MEDICAL ATTENTION IN H A LA T IO N : Move affected person from exposure. If recovery not rapid or complete seek medical attention. If breathing stops, provide artificial respiration. Keep affected person warm and at rest. IN G E ST IO N: DO NOT INDUCE VOMITING. In case of spontaneous vomiting, be sure that vomit can freely drain because of danger of suffocation. Only when conscious, rinse mouth with plenty of water and give plenty of water to drink - (approx 500ml). Keep patient at rest and obtain medical attention. SKIN: Remove contaminated clothing. Wash affected area with plenty of soap and water. If 1/5 10178 - IODOFORM COMPOUND PAINT symptoms occur or persist, obtain medical attention. Launder clothing before re-use. EYES: Rinse immediately with plenty of water for at least 5 minutes while lifting the eye lids. -

Haloform Reaction - Wikipedia
6/13/2020 Haloform reaction - Wikipedia Haloform reaction The haloform reaction is a chemical reaction where a haloform Haloform reaction (CHX , where X is a halogen) is produced by the exhaustive 3 Named after Adolf Lieben halogenation of a methyl ketone (RCOCH3, where R can be either a hydrogen atom, an alkyl or an aryl group), in the presence of a Reaction type Substitution base.[1][2][3] The reaction can be used to transform acetyl groups into reaction carboxyl groups or to produce chloroform (CHCl3), bromoform Identifiers (CHBr3), or iodoform (CHI3) and also cyanide. Organic haloform-reaction Chemistry Portal Contents Mechanism Scope Applications Laboratory scale Industrially As a by-product of water chlorination History References Mechanism In the first step, the halogen disproportionates in the presence of hydroxide to give the halide and hypohalite (example with bromine, but reaction is the same in case of chlorine and iodine; one should only substitute Br for Cl or I): If a secondary alcohol is present, it is oxidized to a ketone by the hypohalite: If a methyl ketone is present, it reacts with the hypohalite in a three-step process: 1. Under basic conditions, the ketone undergoes keto-enol tautomerization. The enolate undergoes electrophilic attack by the hypohalite (containing a halogen with a formal +1 charge). https://en.wikipedia.org/wiki/Haloform_reaction#Iodoform_reaction 1/5 6/13/2020 Haloform reaction - Wikipedia 2. When the α(alpha) position has been exhaustively halogenated, the molecule undergoes a nucleophilic − acyl substitution by hydroxide, with CX3 being the leaving group stabilized by three electron- − withdrawing groups. -

Iodoform Packing
Created by the British Columbia Provincial Nursing Skin and Wound Committee in collaboration with the Wound Clinicians from / Skin and Wound Product Information Sheet Iodoform Packing Classification Antimicrobial/Antiseptic: Iodine Key Points A ravel-resistant strip gauze packing impregnant with Iodoform, an antiseptic effective against pseudomonas. In the presence of exudate, iodoform breaks down to release Iodine (96%) and therefore should be used with caution. The packing’s colour may be opaque to yellowish and may have an oily appearance, as a small amount of oil may have been use to aid in easier removal of product from the container. Should be packed dry. Indications Use under the direction of a Physician, NP, NSWOC/Wound Clinician: o For wounds with or without undermining, sinus tract/tunnels which have signs and symptoms (S&S) of, or are at risk for, local wound infection. Precautions May cause some discomfort/pain, especially when first applied. Avoid using before and after radio-iodine diagnostic tests Make Physician/NP aware of Iodine usage for clients: o Taking lithium, as Iodine may increase the possibility of hypothyroidism when used in combination with lithium. Blood work should be monitored on a regular basis. o With renal impairment, as poor renal function is thought to be a factor in increased iodine levels in serum and urine with prolonged use and use in large wounds. o With thyroid disorders, as they are more susceptible to thyroid metabolism changes in long-term therapy. Thyroid function should be monitored if large areas are being treated for a prolonged period of time. -

Jamaludin Al Anshori, M.Sc. Laboratory of Organic Chemistry
LABORATORY MANUAL OF EXPERIMENTAL ORGANIC CHEMISTRY I Compiled By: Jamaludin Al Anshori, M.Sc. Laboratory of Organic Chemistry Faculty of Mathematics and Natural Sciences Universitas Padjadjaran 2008 LABORATORY MANUAL OF EXPERIMENTAL ORGANIC CHEMISTRY I Compiled by: Jamaludin Al Anshori, M.Sc. LABORATORY OF ORGANIC CHEMISTRY FACULTY OF MATHEMATICS AND NATURAL SCIENCES UNIVERSITAS PADJADJARAN JATINANGOR JATINANGOR, AUGUST 21, 2008 Approved by: Compiler: Head of Organic Chemistry Laboratory Tati Herlina, M.Si. Jamaludin Al Anshori, M.Sc. NIP. 131 772 457 NIP. 132 306 074 i TABLE OF CONTENTS Table of Contents ....................................................................................................................i Preface ................................................................................................................................ iv A. Laboratory’s Rules………………………………………………………………………… v B. Introduction to The Laboratory……………………………………………………………vii Laboratory Techniques : Experiment 1 I.1 Extraction........................................................................................................................1 I.1.1 Introduction........................................................................................................... 1 I.1.2 Using the separating funnel.................................................................................. 2 I.1.3 Procedure ............................................................................................................. 3 I.1.4 Question .............................................................................................................. -

Iodoform: Summary Report
Iodoform: Summary Report Item Type Report Authors Yoon, SeJeong; Gianturco, Stephanie L.; Pavlech, Laura L.; Storm, Kathena D.; Yuen, Melissa V.; Mattingly, Ashlee N. Publication Date 2020-01 Keywords Iodoform; Compounding; Food, Drug, and Cosmetic Act, Section 503B; Food and Drug Administration; Outsourcing facility; Drug compounding; Legislation, Drug; United States Food and Drug Administration Rights Attribution-NoDerivatives 4.0 International Download date 24/09/2021 08:41:01 Item License http://creativecommons.org/licenses/by-nd/4.0/ Link to Item http://hdl.handle.net/10713/12154 Summary Report Iodoform Prepared for: Food and Drug Administration Clinical use of bulk drug substances nominated for inclusion on the 503B Bulks List Grant number: 2U01FD005946 Prepared by: University of Maryland Center of Excellence in Regulatory Science and Innovation (M-CERSI) University of Maryland School of Pharmacy January 2020 This report was supported by the Food and Drug Administration (FDA) of the U.S. Department of Health and Human Services (HHS) as part of a financial assistance award (U01FD005946) totaling $2,342,364, with 100 percent funded by the FDA/HHS. The contents are those of the authors and do not necessarily represent the official views of, nor an endorsement by, the FDA/HHS or the U.S. Government. 1 Table of Contents REVIEW OF NOMINATION ..................................................................................................... 4 METHODOLOGY ................................................................................................................... -
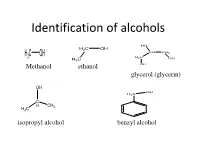
Identification of Alcohols
Identification of alcohols Methanol ethanol glycerol (glycerin) isopropyl alcohol benzyl alcohol Pharmaceutical application of alcohols 1-Methanol, ethanol are very important organic solvents in organic synthesis reactions and crystalization 2- ethanol is used externally as antiseptic for wounds at concentration 70%, in wine preparation,perfumes 3- Benzyl alcohol is used as preservatives in pharmaceutical preparations 4-glyerol is used as wax softener for ears, viscosity modifier & sweetening agent in syrups and levigation of hydrophobic powder in preparation of suspension Identification of alcohols A- Physical Properties: 1- State: liquid 2- color: colorless 3-odour: ethanol, methanol(alcoholic odour) isopropyl alcohol,benzyl alc. (characetristic odor) glycerol (odourless) 4- Miscibility miscible with water except benzyl alcohol which is immiscible and heavier than water 5- Action on litmus paper: neutral B-chemical properties Test Observation Result 1- Acidity test: No eff. ,so it is not acidic compound 2- Dry ignition: a) inflammable with non smoky ,so it is an aliphatic flame (with all except benzyl compound alcohol) OR ,so it is an aromatic b)Inflammable with smoky flame compound 3-Schif’s test: No immediate magenta color ,so it is not aldehhyde 4- Fehling Test: No change in color ,so it is not aldehhyde Test Observation Result 5-Sodium nitroprusside No Blood red color ,so it is not methyl ketone test 6- Oxidation test with Orange color turns to ,so it is an aliphatic potassium dichromate: Green color alcohol For aliphatic alcohol only 1 ml unk. + 1 ml K2 Cr2 O7 +1 ml dil .H2SO4 And heat in BWB for 3 min>>> Orange color turns to Green color then a) Add solid sod carbonate if Eff. -

Food Periodic Table
Food Chemistry Periodic Table Celebrating the International Year of the Periodic Table 2019 Created by Jane K Parker Acknowledgements and thanks to the RSC Food Group Committee: Robert Cordina, Bryan Hanley, Taichi Inuit, John Points, Kathy Ridgway, Martin Rose, Wendy Russell, Mike Saltmarsh, Maud Silvent, Clive Thomson, Kath Whittaker, Pete Wilde, and to Martin Chadwick, Cian Moloney and Ese Omoaruhke, for contributions to the elements, to Flaticons for use of their free icons, and to Alinea and TDMA for photographs of He and Ti. In slideshow mode, click on an element in periodic table to find out more, return via the RSC Food Group Logo. Contact: [email protected] Food Chemistry Periodic Table H He Li Be B C N O F Ne Na Mg Al Si P S Cl Ar K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe Cs Ba La Hf Ta W Re Os Ir Pt Au Hg Ti Pb Bi Po At Rn Fr Ra Ac Rf Db Sg Bh Hs Mt Ds Rg Cn Nh Fl Mc Lv Ts Og H Hydrogen 1 Occurrences in food Roles in food • Core element in organic compounds (fats, proteins, • H+ gives one of the five basic tastes – sour. carbohydrates, vitamins). • the higher the concentration of H+, the lower the pH • OH pH 2 Lemon juice (very sour) • H2 H2 H2 H2 H2 pH 3 Apple (sour) C C C C C C • pH 5 Meat (not sour) H3C C C C C C O H H H H H 2 2 2 2 2 • pH 7 Tea or water (not sour) • Hydrogen-bonds, one of the strongest forms of bonding, are crucial for the 3D-structure of many • Key element in water, H2O, which is 70% of the human body and 70% of many foods. -

Identification of an Unknown – Alcohols, Aldehydes, and Ketones
Identification of an Unknown: Alcohols, Aldehydes, and Ketones How does one determine the identity and structure of an unknown compound? This is not a trivial task. Modern x-ray and spectroscopic techniques have made the job much easier, but for some very complex molecules, identification and structure determination remains a challenge. In addition to spectroscopic information and information obtained from other instrumental methods, chemical reactions can provide useful structural information, and physical properties can contribute significantly to confirming the identity of a compound. In this experiment, you will be asked to identify an unknown liquid, which will be either an alcohol, aldehyde, or ketone. Identification will be accomplished by carrying out chemical tests, called classification tests, preparing a solid derivative of the unknown and determining its melting point (MP), making careful observations, and analyzing the NMR spectrum of the unknown. the carbonyl group O O R OH R H R R alcohol aldehyde ketone A list of alcohols, aldehydes, and ketones, along with the MP of a solid derivative of each compound, is posted on the website. The unknown will be one of these listed compounds. If one can determine to which functional group class (alcohol, aldehyde, or ketone) the unknown belongs, two of the three lists need not be considered and the task will be greatly simplified. To accomplish this, classification tests will be carried out. First, consider some general ways in which alcohols, aldehydes, and ketones react. 1 CLASSIFICATION TESTS, which are simple chemical reactions that produce color changes or form precipitates, can be used to differentiate alcohols, aldehydes, and ketones, and also to provide further structural information.