Variable Character of OOO and MOO Bonding in Side-On
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Buying a Mobile Phone
Buying a mobile phone Getting started A SIM card is the small chip that goes into mobile phones allowing the phone to connect to the local network. Making calls in the UK using your own international SIM card is likely to be expensive so you might want to buy a new SIM or buy another mobile phone with a SIM included. It can sometimes be cheaper to buy an international calling card that will let you make calls home from a landline, mobile phone or phone box. You can buy calling cards from the newsagent’s shop opposite the Parkinson Building. Currently, when using a UK SIM, you will not be charged extra fees to use your UK allowance of minutes, texts or data plan when in countries within the European Economic Area (EEA). Some providers may also have offers for usage in other countries such as the US, so look out for this. There are two different ways to buy a mobile phone: pay-as-you-go or a contract. Please read the following information carefully to see what you will need to get started. Pay-as-you-go You can get a pay-as-you-go mobile phone or SIM card very quickly and it is easy to keep track of how much you are spending on calls. You can buy credit online, in supermarkets, newsagents, petrol stations and at some ATMs. You will also find a free pay-as-you-go SIM card for Lebara mobile in your Welcome Pack that includes £1 pre-loaded credit. You may be able to buy a SIM card in the UK and use it in your own phone from home. -

Southwest Retort
SOUTHWEST RETORT SIXTY-NINTH YEAR OCTOBER 2016 Published for the advancement of Chemists, Chemical Engineers and Chemistry in this area published by The Dallas-Fort Worth Section, with the cooperation of five other local sections of the American Chemical Society in the Southwest Region. Vol. 69(2) OCTOBER 2016 Editorial and Business Offices: Contact the Editor for subscription and advertisement information. Editor: Connie Hendrickson: [email protected] Copy Editor: Mike Vance, [email protected] Business Manager: Danny Dunn: [email protected] The Southwest Retort is published monthly, September through May, by the Dallas-Ft. Worth Section of the American Chemical Society, Inc., for the ACS Sections of the Southwest Region. October 2016 Southwest RETORT 1 TABLE OF CONTENTS Employment Clearing House………….......3 Fifty Years Ago……………………….….....6 ARTICLES and COLUMNS Schulz Award Winner Gale Hunt………….7 And Another Thing……………………….11 Around the Area………………………….14 Letter from the Editor….…..……….........17 SPECIAL EVENTS National Chemistry Week…………………9 NEWS SHORTS Former pesticide ingredient found in dolphins, birds and fish……………………8 Coffee-infused foam removes lead from contaminated water………………………10 Snake venom composition could be related to hormones and diet……………………..13 Detecting blood alcohol content with an electronic skin patch………………...……16 INDEX OF ADVERTISERS Huffman Laboratories……………....……..4 Contact the DFW Section Vance Editing…..…………….…….……….4 General: [email protected] UT Arlington………………………………..4 Education: [email protected] ANA-LAB……………………...….…...……5 Elections: [email protected] Facebook: DFWACS Twitter: acsdfw October 2016 Southwest RETORT 2 EMPLOYMENT CLEARING HOUSE Job applicants should send name, email, and phone, along with type of position and geographical area desired; employers may contact job applicants directly. -
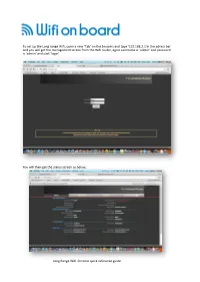
Long Range Wifi Tube Settings
To set up the Long range Wifi, open a new ‘Tab’ on the browers and type ‘192.168.2.1’in the adress bar and you will get the management screen from the Wifi router, again username is ‘admin’ and password is ‘admin’ and click ‘login’ You will then get the status screen as below, Long Range Wifi Chrome quick reference guide Click ‘Easy Setup and select the WAN connections and ‘Client Router Mode’ and click on ‘Next’ On the next screen click ‘Site Survey” Long Range Wifi Chrome quick reference guide This will bring up a screen with all the available WiFi hotspots in the area. Select the WiFi Hotspot you require, the system will connect to most Wifi access points with a signal greater than -80dB ie -79dB to 0dB. Select the Wifi you want in this case BT with FON and click ‘Select’ N o w If you have selected a known Wifi with and access code you will be asked to enter this as the ‘Passkey’ So if you have been to a Bar and have the code this is where you enter it. Set the power level to 24-27 if requires some units do not have this field.. Long Range Wifi Chrome quick reference guide Select Next on the following screens until you reach done and the unit will reboot. Open another tab on you browser and you shold be connected to the Internet, you can also log back into the Long ranage WiFi on 192.168.2.1 and check the status of the connection If you are connected as in this case to BT Openzone, enter your account details and connect to the internet, all other devices will not need to logon to BT or the Public Wifi provider. -

Mobile: the Relationship Channel (Version 4.4) the MMA Would Like to Thank Its Member Sponsors for Their Support in Making This Publication Possible
Mobile: The Relationship Channel (version 4.4) The MMA would like to thank its member sponsors for their support in making this publication possible. Contents Foreword 1 1 Introduction & Purpose 2 2 Why mobile is being used? (The unique role that mobile plays) 4 3 The role that mobile plays in loyalty / The benefits of mobile CRM 13 4 Enablers of mobile loyalty 18 5 Current Mobile Loyalty Landscape 21 5.1 The Current Landscape 5.2 Mobile Wallet 6 Best Practices 31 7 Top Metrics for mobile loyalty programmes 48 8 Barriers to adoption 51 9 Conclusion 54 Foreword by Paul Berney The aim of this White Paper is to give an overview of the current state of the role of mobile in loyalty, with a sister paper focussed more on the like future role that mobile will play set to be published by the MMA in 2014. As this White Paper will demonstrate, although it is early days for the use of mobile in loyalty programmes, there are already some stand-out successes. Both the mobile channel and mobile technologies are having a significant effect on the way that brands engage with the customers. In this way, mobile is both causing and enabling a change in consumer behaviour and the way we interact. The paper will show that mobile can both enhance and extend current loyalty and CRM programmes and at some near future point, mobile will start to replace other channels as consumers become move to a ‘mobile first’ world. As ever the MMA is grateful for the support of its members in helping create this document, in particular the contributing sponsor companies of Advice Group, Aimia, Gemalto, IMI Mobile, Lumata and Velti. -

Ysgol Hendrefelin Headteacher: Mr
Ysgol Hendrefelin Headteacher: Mr. L. Lewis Deputy Head Bryncoch Site: Mr. R. Duford Deputy Head Velindre & Theodore Road Site: Mr. N Lloyd Dear Families, Broadband Connectivity at Home During this time of home learning the access to the internet is essential for all pupils. To help families without broadband connectivity at home the Welsh Government launched a scheme to support some pupils to have increased data via their mobile telephone operator and as a school we have been asked to share this information with you. The scheme is in conjunction with the main UK mobile network operators, namely BT, EE, Three, O2, Vodaphone and Smarty. If successful, the data allowance will be increased to allow pupils to access online educational resources until the 31st July 2021. There are specific conditions for each of the operators: BT Mobile/EE Uplifts are available to both pay as you go and monthly contract customers. Families must have been a customer for at least 3 months. Data allowance to be uplift to unlimited data. Three and SMARTY Uplifts are available to both pay as you go and monthly contract customers. Data allowance to be uplift to unlimited data. Vodaphone Uplifts are available to both pay as you go and monthly contract customers. However, pay as you go customers will need to make a minimum £10 monthly contract. Families must have been a customer for at least 3 months. Data allowance to be uplift to unlimited data. O2 Uplifts are available to both pay as you go and monthly contract customers. Families must have been a customer for at least 6 months. -

Investigation of Lithium Superoxide-Based Batteries
Investigation of Lithium Superoxide-Based Batteries P.I.: K. Amine L. Curtiss Argonne National Laboratory DOE merit review June 1-4, 2020 Project ID# BAT-431 This presentation does not contain any proprietary, confidential, or otherwise restricted information. Overview Timeline Barriers ▪ Start: 2019 ▪ Barriers addressed ▪ Finish: 2023 – Cycle life ▪ 60 % – Capacity – Efficiency Budget Partners • Total project funding • Interactions/ collaborations – DOE share: $ 1280 K • M. Asadi, IIT – Contractor 0 • S. Al-Hallaj and B. Chaplin, UIC • FY 18: $ 400 K • J. G. Wen, ANL • FY 19: $ 450 K • K. C. Lau University of • FY 20: $ 430 K California-Northridge • M. Asadi, IIT 2 Project Objectives and Relevance ▪ Investigation of Li-O2 batteries based on lithium superoxide to achieve understanding of discharge chemistry and how to enhance cycle life. ▪ Use an integrated approach based on experimental synthesis and state-of-the-art characterization combined with high level computational studies focused on materials design and understanding ▪ Li-air batteries based on lithium superoxide have the potential for being the basis for closed systems without need for an external O2 source 3 Milestones Month/ Milestones Year Characterization of electronic properties of high temperature Jun/20 synthesized Ir3Li, (Completed) Voltage profiles and characterization of discharge product by titration Sep/20 and other techniques in an Ir3Li cell, (Completed) TEM studies of large LiO particles, investigation of stability and Dec/21 2 formation mechanism, (Completed) -

CMA Request for Referral Three
HUTCHISON 3G UK INVESTMENTS LIMITED/TELEFONICA EUROPE PLC (the Notified Concentration) REQUEST PURSUANT TO ARTICLE 9(2) OF COUNCIL REGULATION (EU) 139/2004 COMP/M.7612 INTRODUCTION 1. This submission is provided by the United Kingdom’s (UK’s) Competition and Markets Authority (CMA) to the European Commission (the Commission) in support of the CMA’s request made under Article 9(2) of Council Regulation 139/2004 (EUMR) by letter of 2 October 2015, that the Commission refer the whole of the Notified Concentration to the UK so that the CMA can examine the Notified Concentration under the UK merger control provisions in the Enterprise Act 2002.1 TIMING 2. The CMA received a copy of the parties’ Form CO on 14 September 2015. The 15 working day deadline in which to make a request under Article 9 is therefore 5 October 2015. THE UNDERTAKINGS CONCERNED 3. The acquiring company, Hutchison 3G UK Investments Limited (H3G UK) is an indirect wholly owned subsidiary of CK Hutchison Holdings Limited (CKHH) which also owns Hutchison 3G UK Limited (Three), a mobile network operator (MNO) in the UK. Three will become a wholly owned subsidiary of Hutchison 3G UK Investments Limited prior to completion of the Notified Concentration. Three is the most recent MNO entrant on the UK market, and launched its commercial operations on 3 March 2003 with a 2100 MHz 3G 1 The CMA would have jurisdiction under the Enterprise Act 2002 to examine the Notified Concentration were it to be referred back under Article 9 EUMR as the Notified Concentration would constitute a relevant merger situation and the UK turnover of the target exceeds £70 million. -

02 1 Corporate Identity 06.Pdf
our soul telefónica in 2006 02 02·1 Corporate Identity 60 02·2 Business Principles 64 02·3 Corporate Responsibility 68 02·4 Philanthropy 90 02·5 Corporate Governance 96 Telefónica in 2006 02.1 Corporate Identity Telefónica’s Vision delimits and defines the Group’s brand strategy. For this reason, the Group's brand strategy pursues a double objective. On the one hand, to build its institutional profile in order to transfer the value of its brand, Telefónica, as a world leaders in telecommunications. And on the other hand, to configure our offering through our commercial brands (Movistar, O2, Terra) and a wide range of products (duo, trio, imagenio, speedy, superquince, etc.) In order to achieve these two objectives, Telefónica has created a Telefónica's Role system to link and express the portfolio of the group's brands: The Telefónica Master Brand provides the binomial stature: the brand "family system". financial solvency, management capacity, leadership, international projection, credibility, solidity and the know-how of The family system one of the major telecommunication groups in the world. The family system organizes the relationship of the binomial Therefore: made up by the Telefónica brand (Master Brand) and by the • Telefónica is the primary brand for all the Group businesses Group's commercial brands (especially Movistar, O2 and Terra) to create a positive pairing. This system is characterized by: • The values and positioning of the Telefónica brand are the starting point for all Group relationships and communication • The definition of the brand roles. • Telefónica is the only valid speaker from an institutional • The definition of the commercial brand icons or symbols. -

Global Lithium Sources—Industrial Use and Future in the Electric Vehicle Industry: a Review
resources Review Global Lithium Sources—Industrial Use and Future in the Electric Vehicle Industry: A Review Laurence Kavanagh * , Jerome Keohane, Guiomar Garcia Cabellos, Andrew Lloyd and John Cleary EnviroCORE, Department of Science and Health, Institute of Technology Carlow, Kilkenny, Road, Co., R93-V960 Carlow, Ireland; [email protected] (J.K.); [email protected] (G.G.C.); [email protected] (A.L.); [email protected] (J.C.) * Correspondence: [email protected] Received: 28 July 2018; Accepted: 11 September 2018; Published: 17 September 2018 Abstract: Lithium is a key component in green energy storage technologies and is rapidly becoming a metal of crucial importance to the European Union. The different industrial uses of lithium are discussed in this review along with a compilation of the locations of the main geological sources of lithium. An emphasis is placed on lithium’s use in lithium ion batteries and their use in the electric vehicle industry. The electric vehicle market is driving new demand for lithium resources. The expected scale-up in this sector will put pressure on current lithium supplies. The European Union has a burgeoning demand for lithium and is the second largest consumer of lithium resources. Currently, only 1–2% of worldwide lithium is produced in the European Union (Portugal). There are several lithium mineralisations scattered across Europe, the majority of which are currently undergoing mining feasibility studies. The increasing cost of lithium is driving a new global mining boom and should see many of Europe’s mineralisation’s becoming economic. The information given in this paper is a source of contextual information that can be used to support the European Union’s drive towards a low carbon economy and to develop the field of research. -

Charging Ahead FINDING INVESTMENT OPPORTUNITIES from the GREAT BATTERY TAKE-OFF
November 2016 Charging ahead FINDING INVESTMENT OPPORTUNITIES FROM THE GREAT BATTERY TAKE-OFF Duncan Goodwin Alex Scott Head of Global Resources Equities Investment Analyst, Barings, London Baring Global Resources Equities Barings, London Barings | 155 Bishopsgate | London | EC2M 3XY Authorised and Regulated by the Financial Conduct Authority Tel: +44 (0)20 7628 6000 | Fax: +44 (0)20 7638 7928 | www.barings.com FOR INSTITUTIONAL INVESTORS / PROFESSIONAL ADVISERS ONLY Contents EXECUTIVE SUMMARY 3 THE ECONOMICS BEHIND THE TAKE-OFF 4 BATTERY MANUFACTURING: COMPETITION AND COMMODITISATION 6 THE TECHNOLOGY OF TOMORROW 8 CATHODE MANUFACTURERS 13 SEPARATOR MANUFACTURERS 16 COMMODITIES 19 CONCLUSION 23 Barings | 155 Bishopsgate | London | EC2M 3XY November 2016 Tel: +44 (0)20 7628 6000 | Fax: +44 (0)20 7638 7928 | www.barings.com For institutional investors / professional advisers only 2 Executive Summary Following on from the work presented in our white paper In this white paper, we delve deeper into the supply chain “Enabling a Revolution”, this paper is intended as an update on to further identify the potential winners from this structural the sector, and the continuing work we have undertaken. growth theme. We remain permanently cognisant of both the potential for disruptive technology to be, itself, disrupted, as Over the past nine months the idea that we are on the verge of well as for a bubble to emerge within such nascent thematic an electric vehicle take-off has become increasingly palpable. trends. We identify the next rung of potential winners, and also Tesla announced the launch of its long-awaited Model 3 at the re-examine the investment case for those stocks we previously end of March 2016 and already has a pre-order book of more identified as beneficiaries. -

Peroxides, Su Peroxides, and Ozonides of Alkali and Alkaline Earth Metals
Peroxides, Su peroxides, and Ozonides of Alkali and Alkaline Earth Metals Il'ya Ivanovich Vol'nov Head, Laboratory of Peroxide Chemistry N. S. Kurnakov Institute of General and Inorganic Chemistry Academy of Sciences of the USSR, Moscow Translated from Russian by J. Woroncow Life Sciences Group General Dynamics/Convair Division San Diego, California Edited by A. W. Petrocelli Chief, Chemistry and Chemical Engineering Section General Dynamics / Electric Boat Division Groton, Connecticut PLENUM PRESS· NEW YORK· 1966 Born in 1913, Il'ya Ivanovich Vol'nov is head of the laboratory of peroxide chemistry of the N. S. Kurnakov Institute of General and Inorganic Chem istry of the Academy of Sciences of the USSR in Moscow. He joined the Institute in 1939 and since 1949 he has authored more than 50 articles dealing with the chemistry of the inorganic peroxides, superoxides, and ozonides. Vol'nov served as editor for the proceedings of the 2nd All-Union Conference on the Chemistry of Peroxide Compounds, published by the Academy of Sciences in 1963. He was also editor of T. A. Dobrynina's monograph on Lithium Peroxide, published in 1964, and edited a biblio graphical index covering the world-wide literature for the period 1956 to 1962 on the chemistry of peroxide compounds ( other than hydrogen peroxide) published under the auspices of the library of the Academy of Sciences of the USSR. ISBN 978-1-4684-8254-6 ISBN 978-1-4684-8252-2 (eBook) DOl 10.10071978-1-4684-8252-2 Library of Congress Catalog Card Number 66-22125 The original Russian text, first published for the N. -

Batteries: Avoiding Oxygen
PUBLISHED: 25 JULY 2016 | ARTICLE NUMBER: 16115 | DOI: 10.1038/NENERGY.2016.115 news & views BATTERIES Avoiding oxygen In the development of lithium–air batteries, managing the phase change between gaseous oxygen and crystalline lithium peroxide is a key challenge. Now, a high-performing sealed battery with an oxygen anion-redox electrode is presented that does not involve any gas evolution. Laurence J. Hardwick nergy storage in the form of capacity of around 550 Ah kg–1, and the cycle shuttle, that is, a redox species that diffuses rechargeable batteries is becoming can be repeated over 100 times. When used in-between both electrodes essentially increasingly important for a range as the positive electrode in a Li-ion cell, a allowing electrons to flow through the E –1 of applications including transportation specific energy of 1,000 Wh kg was shown, electrolyte. A redox shuttle in this example and grid reserves. Recent interest in non- which rivals the Li–O2 cell and outcompetes could be thought of as a ‘chemical short aqueous metal–oxygen batteries, particularly the state-of-the-art Li-ion technology by circuit’. As shown in Fig. 1, the redox 1 – lithium–oxygen (Li–O2), has stemmed a factor of 2.5–3 at the materials level . shuttle (Sh ) is created from the reaction from their high theoretical gravimetric The cell also showed a minor voltage gap between surface exposed LiO2 and the 1 energy densities . In Li–O2 batteries, the of 0.24 V between discharge and charge, electrolyte solvent (ethylene carbonate). – reactant (O2) is not contained within implying possible high roundtrip efficiencies Sh diffuses to the negative electrode where the cell; instead, it enters from outside in operation.