The Iron-Responsive Genome of the Chiton Acanthopleura Granulata
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Some Aspects of the Biology of Three Northwestern Atlantic Chitons
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Spring 1978 SOME ASPECTS OF THE BIOLOGY OF THREE NORTHWESTERN ATLANTIC CHITONS: TONICELLA RUBRA, TONICELLA MARMOREA, AND ISCHNOCHITON ALBUS (MOLLUSCA: POLYPLACOPHORA) PAUL DAVID LANGER University of New Hampshire, Durham Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation LANGER, PAUL DAVID, "SOME ASPECTS OF THE BIOLOGY OF THREE NORTHWESTERN ATLANTIC CHITONS: TONICELLA RUBRA, TONICELLA MARMOREA, AND ISCHNOCHITON ALBUS (MOLLUSCA: POLYPLACOPHORA)" (1978). Doctoral Dissertations. 2329. https://scholars.unh.edu/dissertation/2329 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. INFORMATION TO USERS This material was produced from a microfilm copy of the original document. While the most advanced technological means to photograph and reproduce this document have been used, the quality is heavily dependent upon the quality of the original submitted. The following explanation of techniques is provided to help you understand markings or patterns which may appear on this reproduction. 1.The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting thru an image and duplicating adjacent pages to insure you complete continuity. 2. When an image on the film is obliterated with a large round black mark, it is an indication that the photographer suspected that the copy may have moved during exposure and thus cause a blurred image. -
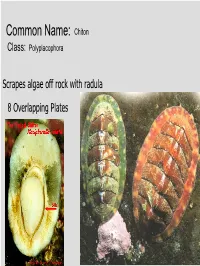
Common Name: Chiton Class: Polyplacophora
Common Name: Chiton Class: Polyplacophora Scrapes algae off rock with radula 8 Overlapping Plates Phylum? Mollusca Class? Gastropoda Common name? Brown sea hare Class? Scaphopoda Common name? Tooth shell or tusk shell Mud Tentacle Foot Class? Gastropoda Common name? Limpet Phylum? Mollusca Class? Bivalvia Class? Gastropoda Common name? Brown sea hare Phylum? Mollusca Class? Gastropoda Common name? Nudibranch Class? Cephalopoda Cuttlefish Octopus Squid Nautilus Phylum? Mollusca Class? Gastropoda Most Bivalves are Filter Feeders A B E D C • A: Mantle • B: Gill • C: Mantle • D: Foot • E: Posterior adductor muscle I.D. Green: Foot I.D. Red Gills Three Body Regions 1. Head – Foot 2. Visceral Mass 3. Mantle A B C D • A: Radula • B: Mantle • C: Mouth • D: Foot What are these? Snail Radulas Dorsal HingeA Growth line UmboB (Anterior) Ventral ByssalC threads Mussel – View of Outer Shell • A: Hinge • B: Umbo • C: Byssal threads Internal Anatomy of the Bay Mussel A B C D • A: Labial palps • B: Mantle • C: Foot • D: Byssal threads NacreousB layer Posterior adductorC PeriostracumA muscle SiphonD Mantle Byssal threads E Internal Anatomy of the Bay Mussel • A: Periostracum • B: Nacreous layer • C: Posterior adductor muscle • D: Siphon • E: Mantle Byssal gland Mantle Gill Foot Labial palp Mantle Byssal threads Gill Byssal gland Mantle Foot Incurrent siphon Byssal Labial palp threads C D B A E • A: Foot • B: Gills • C: Posterior adductor muscle • D: Excurrent siphon • E: Incurrent siphon Heart G F H E D A B C • A: Foot • B: Gills • C: Mantle • D: Excurrent siphon • E: Incurrent siphon • F: Posterior adductor muscle • G: Labial palps • H: Anterior adductor muscle Siphon or 1. -

Marine Invertebrate Field Guide
Marine Invertebrate Field Guide Contents ANEMONES ....................................................................................................................................................................................... 2 AGGREGATING ANEMONE (ANTHOPLEURA ELEGANTISSIMA) ............................................................................................................................... 2 BROODING ANEMONE (EPIACTIS PROLIFERA) ................................................................................................................................................... 2 CHRISTMAS ANEMONE (URTICINA CRASSICORNIS) ............................................................................................................................................ 3 PLUMOSE ANEMONE (METRIDIUM SENILE) ..................................................................................................................................................... 3 BARNACLES ....................................................................................................................................................................................... 4 ACORN BARNACLE (BALANUS GLANDULA) ....................................................................................................................................................... 4 HAYSTACK BARNACLE (SEMIBALANUS CARIOSUS) .............................................................................................................................................. 4 CHITONS ........................................................................................................................................................................................... -

Phylum MOLLUSCA Chitons, Bivalves, Sea Snails, Sea Slugs, Octopus, Squid, Tusk Shell
Phylum MOLLUSCA Chitons, bivalves, sea snails, sea slugs, octopus, squid, tusk shell Bruce Marshall, Steve O’Shea with additional input for squid from Neil Bagley, Peter McMillan, Reyn Naylor, Darren Stevens, Di Tracey Phylum Aplacophora In New Zealand, these are worm-like molluscs found in sandy mud. There is no shell. The tiny MOLLUSCA solenogasters have bristle-like spicules over Chitons, bivalves, sea snails, sea almost the whole body, a groove on the underside of the body, and no gills. The more worm-like slugs, octopus, squid, tusk shells caudofoveates have a groove and fewer spicules but have gills. There are 10 species, 8 undescribed. The mollusca is the second most speciose animal Bivalvia phylum in the sea after Arthropoda. The phylum Clams, mussels, oysters, scallops, etc. The shell is name is taken from the Latin (molluscus, soft), in two halves (valves) connected by a ligament and referring to the soft bodies of these creatures, but hinge and anterior and posterior adductor muscles. most species have some kind of protective shell Gills are well-developed and there is no radula. and hence are called shellfish. Some, like sea There are 680 species, 231 undescribed. slugs, have no shell at all. Most molluscs also have a strap-like ribbon of minute teeth — the Scaphopoda radula — inside the mouth, but this characteristic Tusk shells. The body and head are reduced but Molluscan feature is lacking in clams (bivalves) and there is a foot that is used for burrowing in soft some deep-sea finned octopuses. A significant part sediments. The shell is open at both ends, with of the body is muscular, like the adductor muscles the narrow tip just above the sediment surface for and foot of clams and scallops, the head-foot of respiration. -

The Malacological Society of London
ACKNOWLEDGMENTS This meeting was made possible due to generous contributions from the following individuals and organizations: Unitas Malacologica The program committee: The American Malacological Society Lynn Bonomo, Samantha Donohoo, The Western Society of Malacologists Kelly Larkin, Emily Otstott, Lisa Paggeot David and Dixie Lindberg California Academy of Sciences Andrew Jepsen, Nick Colin The Company of Biologists. Robert Sussman, Allan Tina The American Genetics Association. Meg Burke, Katherine Piatek The Malacological Society of London The organizing committee: Pat Krug, David Lindberg, Julia Sigwart and Ellen Strong THE MALACOLOGICAL SOCIETY OF LONDON 1 SCHEDULE SUNDAY 11 AUGUST, 2019 (Asilomar Conference Center, Pacific Grove, CA) 2:00-6:00 pm Registration - Merrill Hall 10:30 am-12:00 pm Unitas Malacologica Council Meeting - Merrill Hall 1:30-3:30 pm Western Society of Malacologists Council Meeting Merrill Hall 3:30-5:30 American Malacological Society Council Meeting Merrill Hall MONDAY 12 AUGUST, 2019 (Asilomar Conference Center, Pacific Grove, CA) 7:30-8:30 am Breakfast - Crocker Dining Hall 8:30-11:30 Registration - Merrill Hall 8:30 am Welcome and Opening Session –Terry Gosliner - Merrill Hall Plenary Session: The Future of Molluscan Research - Merrill Hall 9:00 am - Genomics and the Future of Tropical Marine Ecosystems - Mónica Medina, Pennsylvania State University 9:45 am - Our New Understanding of Dead-shell Assemblages: A Powerful Tool for Deciphering Human Impacts - Sue Kidwell, University of Chicago 2 10:30-10:45 -

Download Preprint
1 Mobilising molluscan models and genomes in biology 2 Angus Davison1 and Maurine Neiman2 3 1. School of Life Sciences, University Park, University of Nottingham, NG7 2RD, UK 4 2. Department of Biology, University of Iowa, Iowa City, IA, USA and Department of Gender, 5 Women's, and Sexuality Studies, University of Iowa, Iowa, City, IA, USA 6 Abstract 7 Molluscs are amongst the most ancient, diverse, and important of all animal taxa. Even so, 8 no individual mollusc species has emerged as a broadly applied model system in biology. 9 We here make the case that both perceptual and methodological barriers have played a role 10 in the relative neglect of molluscs as research organisms. We then summarize the current 11 application and potential of molluscs and their genomes to address important questions in 12 animal biology, and the state of the field when it comes to the availability of resources such 13 as genome assemblies, cell lines, and other key elements necessary to mobilising the 14 development of molluscan model systems. We conclude by contending that a cohesive 15 research community that works together to elevate multiple molluscan systems to ‘model’ 16 status will create new opportunities in addressing basic and applied biological problems, 17 including general features of animal evolution. 18 Introduction 19 Molluscs are globally important as sources of food, calcium and pearls, and as vectors of 20 human disease. From an evolutionary perspective, molluscs are notable for their remarkable 21 diversity: originating over 500 million years ago, there are over 70,000 extant mollusc 22 species [1], with molluscs present in virtually every ecosystem. -

(Approx) Mixed Micro Shells (22G Bags) Philippines € 10,00 £8,64 $11,69 Each 22G Bag Provides Hours of Fun; Some Interesting Foraminifera Also Included
Special Price £ US$ Family Genus, species Country Quality Size Remarks w/o Photo Date added Category characteristic (€) (approx) (approx) Mixed micro shells (22g bags) Philippines € 10,00 £8,64 $11,69 Each 22g bag provides hours of fun; some interesting Foraminifera also included. 17/06/21 Mixed micro shells Ischnochitonidae Callistochiton pulchrior Panama F+++ 89mm € 1,80 £1,55 $2,10 21/12/16 Polyplacophora Ischnochitonidae Chaetopleura lurida Panama F+++ 2022mm € 3,00 £2,59 $3,51 Hairy girdles, beautifully preserved. Web 24/12/16 Polyplacophora Ischnochitonidae Ischnochiton textilis South Africa F+++ 30mm+ € 4,00 £3,45 $4,68 30/04/21 Polyplacophora Ischnochitonidae Ischnochiton textilis South Africa F+++ 27.9mm € 2,80 £2,42 $3,27 30/04/21 Polyplacophora Ischnochitonidae Stenoplax limaciformis Panama F+++ 16mm+ € 6,50 £5,61 $7,60 Uncommon. 24/12/16 Polyplacophora Chitonidae Acanthopleura gemmata Philippines F+++ 25mm+ € 2,50 £2,16 $2,92 Hairy margins, beautifully preserved. 04/08/17 Polyplacophora Chitonidae Acanthopleura gemmata Australia F+++ 25mm+ € 2,60 £2,25 $3,04 02/06/18 Polyplacophora Chitonidae Acanthopleura granulata Panama F+++ 41mm+ € 4,00 £3,45 $4,68 West Indian 'fuzzy' chiton. Web 24/12/16 Polyplacophora Chitonidae Acanthopleura granulata Panama F+++ 32mm+ € 3,00 £2,59 $3,51 West Indian 'fuzzy' chiton. 24/12/16 Polyplacophora Chitonidae Chiton tuberculatus Panama F+++ 44mm+ € 5,00 £4,32 $5,85 Caribbean. 24/12/16 Polyplacophora Chitonidae Chiton tuberculatus Panama F++ 35mm € 2,50 £2,16 $2,92 Caribbean. 24/12/16 Polyplacophora Chitonidae Chiton tuberculatus Panama F+++ 29mm+ € 3,00 £2,59 $3,51 Caribbean. -

<I>Acanthopleura Gemmata</I>
NOTES 339 Mauri, M. and E. Orlando. 1983. Variability of zinc and manganese concentrations in relation to sex and season in the bivalve Donax trunculus. Mar. Pollut. Bull. 14: 342-346. McConchie, D. and L. M. Lawrance. 1991. The origin of high cadmium loads in some bivalves molluscs from Shark Bay, Western Australia: a new mechanism for cadmium uptake by filter feeding organisms. Arch. Environ. Contam. Toxicol. 21: 303-310. Nugegoda, D. and P. S. Rainbow. 1987. The effect of temperature on zinc regulation by the decapod crustacean Palaemon elegans Rathke. Ophelia 27: 17-30. Orlando, E. ]985. Valutazione dell'inquinamento marino da metalli pesanti tramite ]'uso di indicatori biologici. Oebalia XI: 93-100. Orren, M. J., G. A. Eagle, H. F.-K. O. Hennig and A. Green. 1980. Variations in trace metal content of the mussel Choromytilus meridionalis (Kr.) with season and sex. Mar. Pollut. Bull. 11: 253- 257. Rainbow, P. S. and A. G. Scott. 1979. Two heavy metal-binding proteins in the midgut gland of the crab Carcinus maenas. Mar. BioI. 55: 143-150. DATE ACCEPTED: April 13, ]994. ADDRESSES: (K.F.) Western Australian Marine Research Laboratories, PO Box 20, North Beach 6020, Australia; (CS.) Chemistry Centre of Western Australia, 125 Hay Street, East Perth 6004, Australia; (M.J.) Health Department of Western Australia, 189 Royal Street, East Perth 6004, Aus- tralia. BULLETINOF MARINESCIENCE,56( I): 339-343, 1995 KARYOLOGICAL STUDIES ON THE COMMON ROCKY EGYPTIAN CHITON, ACANTHOPLEURA GEMMATA (POLYPLACOPHORA: MOLLUSCA) Ahmed E. Yaseen, Abdel-Baset M. Ebaid and I. S. Kawashti Acanthopleura gemmata (Blainville, 1925) is one of the commonest polypla- cophoran species and is very common along the Egyptian coasts (northwestern part of the Red Sea) (Soliman and Habib, 1990). -

Quality Molluscan Genomes Jin Sun 1, Runsheng Li 2, Chong Chen 3, Julia D
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.31.424979; this version posted January 2, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Benchmarking Oxford Nanopore read assemblers for high- quality molluscan genomes Jin Sun 1, Runsheng Li 2, Chong Chen 3, Julia D. Sigwart 4,5, Kevin M. Kocot 6* 1 Institute of Evolution & Marine Biodiversity, Ocean University of China, Qingdao 266003, China 2 Department of Infectious Diseases and Public Health, Jockey Club College of Veterinary Medicine and Life Sciences, City University of Hong Kong, Kowloon, Hong Kong, China 3 X-STAR, Japan Agency for Marine-Earth Science and Technology (JAMSTEC), 2-15 Natsushima-cho, Yokosuka, Kanagawa Prefecture 237-0061, Japan 4 Senckenberg Museum, Frankfurt, Germany 5 Queen’s University Belfast, Marine Laboratory, Portaferry, N Ireland 6 Department of Biological Sciences and Alabama Museum of Natural History, University of Alabama, Tuscaloosa, Alabama, 35487, USA Keywords: Molluscan genomes, assembly, Oxford Nanopore Technology, scaly-foot snail, Mytilus, phylogeny Summary Choosing the optimum assembly approach is essential to achieving a high-quality genome assembly suitable for comparative and evolutionary genomic investigations. Significant recent progress in long-read sequencing technologies such as PacBio and Oxford Nanopore Technologies (ONT) also brought about a large variety of assemblers. Although these have been extensively tested on model species such as Homo sapiens and Drosophila melanogaster, such benchmarking has not been done in Mollusca which lacks widely adopted model species. -

Limpets and Chitons, Teacher Guide
TEACHER BACKGROUND Unit 5 - Limpets and Chitons Limpets and Chitons (Ki tons) Key Concepts 1. Limpets are single shelled marine animals that use a flat, muscular foot to remain attached to rocks. 2. Chitons are marine animals which have eight shell plates for protection and use a flat, muscular foot to remain attached to rocks. 3. Both limpets and chitons use their rasping tongue or radula to graze on tiny algae which covers the rocks in tidepools. Background Limpets and chitons are common and commonly overlooked tidepool animals. We tend to think of free swimming fish and scurrying crabs when we think of marine animals. However, a great many marine animals, such as limpets and chitons, lead a sedentary existence, firmly affixed to the bottom or some other feature. Both limpets and chitons possess a large, muscular foot which they use for attachment. During low tides, most limpets and chitons stay in one spot. When submerged during high tides, they glide slowly over the rocks. As the tide falls, they return to precisely the same spot. For limpets that spot is a neat “scar” the exact size and shape of the limpet. Limpets and chitons eat very small but nevertheless visible (macroscopic) plants (algae). Using a rasping tongue, called a radula, a limpet or chiton grazes the surface scraping off the microscopic plants as it travels. This feeding action underscores the interrelationships that exist between plants and animals. The existence of any species of animal is directly or indirectly related to the existence of some species of plant. Although both molluscs, the shells of limpets and chitons are quite different. -

A Molecular Phylogeny of the Patellogastropoda (Mollusca: Gastropoda)
^03 Marine Biology (2000) 137: 183-194 ® Spnnger-Verlag 2000 M. G. Harasevvych A. G. McArthur A molecular phylogeny of the Patellogastropoda (Mollusca: Gastropoda) Received: 5 February 1999 /Accepted: 16 May 2000 Abstract Phylogenetic analyses of partiaJ J8S rDNA formia" than between the Patellogastropoda and sequences from species representing all living families of Orthogastropoda. Partial 18S sequences support the the order Patellogastropoda, most other major gastro- inclusion of the family Neolepetopsidae within the su- pod groups (Cocculiniformia, Neritopsma, Vetigastro- perfamily Acmaeoidea, and refute its previously hy- poda, Caenogastropoda, Heterobranchia, but not pothesized position as sister group to the remaining Neomphalina), and two additional classes of the phylum living Patellogastropoda. This region of the Í8S rDNA Mollusca (Cephalopoda, Polyplacophora) confirm that gene diverges at widely differing rates, spanning an order Patellogastropoda comprises a robust clade with high of magnitude among patellogastropod lineages, and statistical support. The sequences are characterized by therefore does not provide meaningful resolution of the the presence of several insertions and deletions that are relationships among higher taxa of patellogastropods. unique to, and ubiquitous among, patellogastropods. Data from one or more genes that evolve more uni- However, this portion of the 18S gene is insufficiently formly and more rapidly than the ISSrDNA gene informative to provide robust support for the mono- (possibly one or more -

An Annotated Checklist of the Marine Macroinvertebrates of Alaska David T
NOAA Professional Paper NMFS 19 An annotated checklist of the marine macroinvertebrates of Alaska David T. Drumm • Katherine P. Maslenikov Robert Van Syoc • James W. Orr • Robert R. Lauth Duane E. Stevenson • Theodore W. Pietsch November 2016 U.S. Department of Commerce NOAA Professional Penny Pritzker Secretary of Commerce National Oceanic Papers NMFS and Atmospheric Administration Kathryn D. Sullivan Scientific Editor* Administrator Richard Langton National Marine National Marine Fisheries Service Fisheries Service Northeast Fisheries Science Center Maine Field Station Eileen Sobeck 17 Godfrey Drive, Suite 1 Assistant Administrator Orono, Maine 04473 for Fisheries Associate Editor Kathryn Dennis National Marine Fisheries Service Office of Science and Technology Economics and Social Analysis Division 1845 Wasp Blvd., Bldg. 178 Honolulu, Hawaii 96818 Managing Editor Shelley Arenas National Marine Fisheries Service Scientific Publications Office 7600 Sand Point Way NE Seattle, Washington 98115 Editorial Committee Ann C. Matarese National Marine Fisheries Service James W. Orr National Marine Fisheries Service The NOAA Professional Paper NMFS (ISSN 1931-4590) series is pub- lished by the Scientific Publications Of- *Bruce Mundy (PIFSC) was Scientific Editor during the fice, National Marine Fisheries Service, scientific editing and preparation of this report. NOAA, 7600 Sand Point Way NE, Seattle, WA 98115. The Secretary of Commerce has The NOAA Professional Paper NMFS series carries peer-reviewed, lengthy original determined that the publication of research reports, taxonomic keys, species synopses, flora and fauna studies, and data- this series is necessary in the transac- intensive reports on investigations in fishery science, engineering, and economics. tion of the public business required by law of this Department.