299823 Hamid Ghanavi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rospuda Valley Survey 2007
Rospuda Valley Survey 2007 review of surveyed groups European species lists Biodiversity Survey final report - November 2008 Cite this report as: European Biodiversity Survey (): Biodiversity Survey Rospuda Valley, Final Report. Gronin- gen, European Biodiversity Survey. © European Biodiversity Survey (EBS). is is an open-access publication distributed under the Creative Commons Attribution License which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Photos on cover: top le corner: Nehallenia speciosa, by Tim Faasen. Middle right: Boloria eu- phrosyne, by Tim Faasen. Middle le: Colobochyla salicalis, by Wouter Moerland. Right bottom: Calcereous fen, by Bram Kuijper. European Biodiversity Survey Van Royenlaan A ES Groningen e-mail: info at biodiversitysurvey.eu www: www.biodiversitysurvey.eu is is not a eld guide. e Rospuda Vally and especially its valuable bogs are very vulnerable. ough more information on the distribution of species in the Rospuda Vally is important, please think twice before you enter the area. Contents Preface Introduction . Geography and natural history of the Rospuda area ................. . Pristine character ..................................... . ViaBaltica ......................................... . Vegetation zonation in the mire ............................. . Rationale for this survey ................................. . Methods .......................................... Aquatic fauna . Introduction ....................................... -

SYSTEMATICS of the MEGADIVERSE SUPERFAMILY GELECHIOIDEA (INSECTA: LEPIDOPTEA) DISSERTATION Presented in Partial Fulfillment of T
SYSTEMATICS OF THE MEGADIVERSE SUPERFAMILY GELECHIOIDEA (INSECTA: LEPIDOPTEA) DISSERTATION Presented in Partial Fulfillment of the Requirements for The Degree of Doctor of Philosophy in the Graduate School of The Ohio State University By Sibyl Rae Bucheli, M.S. ***** The Ohio State University 2005 Dissertation Committee: Approved by Dr. John W. Wenzel, Advisor Dr. Daniel Herms Dr. Hans Klompen _________________________________ Dr. Steven C. Passoa Advisor Graduate Program in Entomology ABSTRACT The phylogenetics, systematics, taxonomy, and biology of Gelechioidea (Insecta: Lepidoptera) are investigated. This superfamily is probably the second largest in all of Lepidoptera, and it remains one of the least well known. Taxonomy of Gelechioidea has been unstable historically, and definitions vary at the family and subfamily levels. In Chapters Two and Three, I review the taxonomy of Gelechioidea and characters that have been important, with attention to what characters or terms were used by different authors. I revise the coding of characters that are already in the literature, and provide new data as well. Chapter Four provides the first phylogenetic analysis of Gelechioidea to include molecular data. I combine novel DNA sequence data from Cytochrome oxidase I and II with morphological matrices for exemplar species. The results challenge current concepts of Gelechioidea, suggesting that traditional morphological characters that have united taxa may not be homologous structures and are in need of further investigation. Resolution of this problem will require more detailed analysis and more thorough characterization of certain lineages. To begin this task, I conduct in Chapter Five an in- depth study of morphological evolution, host-plant selection, and geographical distribution of a medium-sized genus Depressaria Haworth (Depressariinae), larvae of ii which generally feed on plants in the families Asteraceae and Apiaceae. -

Lepidoptera of North America 5
Lepidoptera of North America 5. Contributions to the Knowledge of Southern West Virginia Lepidoptera Contributions of the C.P. Gillette Museum of Arthropod Diversity Colorado State University Lepidoptera of North America 5. Contributions to the Knowledge of Southern West Virginia Lepidoptera by Valerio Albu, 1411 E. Sweetbriar Drive Fresno, CA 93720 and Eric Metzler, 1241 Kildale Square North Columbus, OH 43229 April 30, 2004 Contributions of the C.P. Gillette Museum of Arthropod Diversity Colorado State University Cover illustration: Blueberry Sphinx (Paonias astylus (Drury)], an eastern endemic. Photo by Valeriu Albu. ISBN 1084-8819 This publication and others in the series may be ordered from the C.P. Gillette Museum of Arthropod Diversity, Department of Bioagricultural Sciences and Pest Management Colorado State University, Fort Collins, CO 80523 Abstract A list of 1531 species ofLepidoptera is presented, collected over 15 years (1988 to 2002), in eleven southern West Virginia counties. A variety of collecting methods was used, including netting, light attracting, light trapping and pheromone trapping. The specimens were identified by the currently available pictorial sources and determination keys. Many were also sent to specialists for confirmation or identification. The majority of the data was from Kanawha County, reflecting the area of more intensive sampling effort by the senior author. This imbalance of data between Kanawha County and other counties should even out with further sampling of the area. Key Words: Appalachian Mountains, -

Western Ghats), Idukki District, Kerala, India
International Journal of Entomology Research International Journal of Entomology Research ISSN: 2455-4758 Impact Factor: RJIF 5.24 www.entomologyjournals.com Volume 3; Issue 2; March 2018; Page No. 114-120 The moths (Lepidoptera: Heterocera) of vagamon hills (Western Ghats), Idukki district, Kerala, India Pratheesh Mathew, Sekar Anand, Kuppusamy Sivasankaran, Savarimuthu Ignacimuthu* Entomology Research Institute, Loyola College, University of Madras, Chennai, Tamil Nadu, India Abstract The present study was conducted at Vagamon hill station to evaluate the biodiversity of moths. During the present study, a total of 675 moth specimens were collected from the study area which represented 112 species from 16 families and eight super families. Though much of the species has been reported earlier from other parts of India, 15 species were first records for the state of Kerala. The highest species richness was shown by the family Erebidae and the least by the families Lasiocampidae, Uraniidae, Notodontidae, Pyralidae, Yponomeutidae, Zygaenidae and Hepialidae with one species each. The results of this preliminary study are promising; it sheds light on the unknown biodiversity of Vagamon hills which needs to be strengthened through comprehensive future surveys. Keywords: fauna, lepidoptera, biodiversity, vagamon, Western Ghats, Kerala 1. Introduction Ghats stretches from 8° N to 22° N. Due to increasing Arthropods are considered as the most successful animal anthropogenic activities the montane grasslands and adjacent group which consists of more than two-third of all animal forests face several threats (Pramod et al. 1997) [20]. With a species on earth. Class Insecta comprise about 90% of tropical wide array of bioclimatic and topographic conditions, the forest biomass (Fatimah & Catherine 2002) [10]. -

Allium Sativum, Rosmarinus Officinalis, and Salvia Officinalis
insects Article Allium sativum, Rosmarinus officinalis, and Salvia officinalis Essential Oils: A Spiced Shield against Blowflies Stefano Bedini 1 , Salvatore Guarino 2 , Maria Cristina Echeverria 3 , Guido Flamini 4 , Roberta Ascrizzi 4 , Augusto Loni 1 and Barbara Conti 1,* 1 Department of Agriculture, Food and Environment- University of Pisa, via del Borghetto 80, 56126 Pisa, Italy; [email protected] (S.B.); [email protected] (A.L.) 2 Institute of Biosciences and Bioresources (IBBR), National Research Council of Italy (CNR), Corso Calatafimi 414, 90129 Palermo, Italy; [email protected] 3 Facultad de Ingeniería en Ciencias Agropecuarias y Ambientales. Universidad Técnica del Norte, Av 17 de Julio 5-21, Ibarra 100105, Ecuador; [email protected] 4 Department of Pharmacy, University of Pisa, Via Bonanno 6, 56126 Pisa, Italy; guido.fl[email protected] (G.F.); [email protected] (R.A.) * Correspondence: [email protected] Received: 4 February 2020; Accepted: 20 February 2020; Published: 25 February 2020 Abstract: Blowflies are known vectors of many foodborne pathogens and unintentional human ingestion of maggots by meat consumption may lead to intestinal myiasis. In fact, the control of insect pests is an important aspect of industrial and home-made food processing and blowflies (Diptera: Calliphoridae), which are among the most important pests involved in the damage of meat products. Most spices, largely used in food preparations and industry, contain essential oils that are toxic and repellent against insects and exert antimicrobial activity. In this study, we assessed the electro-antennographic responses, the oviposition deterrence, the toxicity, and the repellence of the essential oils (EOs) of Allium sativum L., Salvia officinalis L., and Rosmarinus officinalis L. -

Autographa Gamma
1 Table of Contents Table of Contents Authors, Reviewers, Draft Log 4 Introduction to the Reference 6 Soybean Background 11 Arthropods 14 Primary Pests of Soybean (Full Pest Datasheet) 14 Adoretus sinicus ............................................................................................................. 14 Autographa gamma ....................................................................................................... 26 Chrysodeixis chalcites ................................................................................................... 36 Cydia fabivora ................................................................................................................. 49 Diabrotica speciosa ........................................................................................................ 55 Helicoverpa armigera..................................................................................................... 65 Leguminivora glycinivorella .......................................................................................... 80 Mamestra brassicae....................................................................................................... 85 Spodoptera littoralis ....................................................................................................... 94 Spodoptera litura .......................................................................................................... 106 Secondary Pests of Soybean (Truncated Pest Datasheet) 118 Adoxophyes orana ...................................................................................................... -

Buletinul Известия Journal
Buletinul AŞM. Ştiinţele vieţii. Nr. 3(330) 2016 Buletinul AŞM. Ştiinţele vieţii. Nr. 3(330) 2016 ISSN 1857-064X Categoria B BULETINUL ACADEMIEI DE ŞTIINŢE A MOLDOVEI Ştiinţele vieţii ИЗВЕСТИЯ АКАДЕМИИ НАУК МОЛДОВЫ Науки о жизНи JOURNAL OF ACADEMY OF SCIENCES OF MOLDOVA LIFE SCIENCES 3 (330) 2016 Chişinău Buletinul AŞM. Ştiinţele vieţii. Nr. 3(330) 2016 Buletinul AŞM. Ştiinţele vieţii. Nr. 3(330) 2016 COLEGIUL DE REDACŢIE Redactor-şef Valentina CIOCHINĂ Teodor FURDUI Institutul de Fiziologie şi Sanocreatologie al Aca- Institutul de Fiziologie şi Sanocreatologie al Aca- demiei de Ştiinţe a Moldovei, Laboratorul Fiziolo- demiei de Ştiinţe a Moldovei, Laboratorul Fiziolo- gia stresului, adaptării şi Sanocreatologie generală, gia stresului, adaptării şi Sanocreatologie generală, doctor, conferenţiar. Adresa: str. Academiei, 1, MD- academician, doctor habilitat, profesor. Adresa: str. 2028 Chişinău, Republica Moldova. Tel.: (+373)22 Academiei, 1, MD-2028 Chişinău, Republica Mol- 725152; E-mail: [email protected] dova. Tel.: (+373)22 725209; (+373) 069972538 Gheorghe DUCA Redactor-şef adjunct Institutul de Chimie al Academiei de Ştiinţe a Moldovei, Centrul Chimie Fizică şi Nanocompozite, Ion TODERAŞ academician, doctor habilitat, profesor. Adresa: Institutul de Zoologie al Academiei de Ştiinţe bd.Ştefan cel Mare, 1, MD-2001 Chişinău, a Moldovei, Centrul de Cercetare a Invaziilor Republica Moldova. Tel/Fax.: (+373)22 271478; Biologice, Laboratorul de Sistematică şi Filogenie E-mail: [email protected], [email protected] moleculară, academician, doctor habilitat, profesor. Adresa: str. Academiei, 1, MD-2028 Chişinău, Maria DUCA Republica Moldova. Tel.: (+373)22 731255; Universitatea Academiei de Ştiinţe a Moldovei, E-mail:[email protected] Centrul universitar Genetică funcţională, Secretar responsabil academician, doctor habilitat, profesor. -

Project Update: June 2013 the Monte Iberia Plateau at The
Project Update: June 2013 The Monte Iberia plateau at the Alejandro de Humboldt National Park (AHNP) was visited in April and June of 2013. A total of 152 butterflies and moths grouped in 22 families were recorded. In total, 31 species of butterflies belonging to five families were observed, all but two new records to area (see list below). Six species and 12 subspecies are Cuban endemics, including five endemics restricted to the Nipe-Sagua- Baracoa. In total, 108 species of moths belonging to 17 families were registered, including 25 endemic species of which five inhabit exclusively the NSB Mountains (see list below). In total, 52 butterflies and endemic moth species were photographed to be included in a guide of butterflies and endemic moths inhabiting Monte Iberia. Vegetation types sampled were the evergreen forests, rainforest, and charrascals (scrub on serpentine soil) at both north and southern slopes of Monte Iberia plateau Sixteen butterfly species were observed in transects. Park authorities were contacted in preparation on a workshop to capacitate park staff. Butterfly and moth species recorded at different vegetation types of Monte Iberia plateau in April and June of 2013. Symbols and abbreviations: ***- Nipe-Sagua-Baracoa endemic, **- Cuban endemic species, *- Cuban endemic subspecies, F- species photographed, vegetation types: DV- disturbed vegetation, EF- evergreen forest, RF- rainforest, CH- charrascal. "BUTTERFLIES" PAPILIONIDAE Papilioninae Heraclides pelaus atkinsi *F/EF/RF Heraclides thoas oviedo *F/CH Parides g. gundlachianus **F/EF/RF/CH HESPERIIDAE Hesperiinae Asbolis capucinus F/RF/CH Choranthus radians F/EF/CH Cymaenes tripunctus EF Perichares p. philetes F/CH Pyrginae Burca cubensis ***F/RF/CH Ephyriades arcas philemon F/EF/RF Ephyriades b. -

Comparative Morphology of the Male Genitalia in Lepidoptera
COMPARATIVE MORPHOLOGY OF THE MALE GENITALIA IN LEPIDOPTERA. By DEV RAJ MEHTA, M. Sc.~ Ph. D. (Canta.b.), 'Univefsity Scholar of the Government of the Punjab, India (Department of Zoology, University of Oambridge). CONTENTS. PAGE. Introduction 197 Historical Review 199 Technique. 201 N ontenclature 201 Function • 205 Comparative Morphology 206 Conclusions in Phylogeny 257 Summary 261 Literature 1 262 INTRODUCTION. In the domains of both Morphology and Taxonomy the study' of Insect genitalia has evoked considerable interest during the past half century. Zander (1900, 1901, 1903) suggested a common structural plan for the genitalia in various orders of insects. This work stimulated further research and his conclusions were amplified by Crampton (1920) who homologized the different parts in the genitalia of Hymenoptera, Mecoptera, Neuroptera, Diptera, Trichoptera Lepidoptera, Hemiptera and Strepsiptera with those of more generalized insects like the Ephe meroptera and Thysanura. During this time the use of genitalic charac ters for taxonomic purposes was also realized particularly in cases where the other imaginal characters had failed to serve. In this con nection may be mentioned the work of Buchanan White (1876), Gosse (1883), Bethune Baker (1914), Pierce (1909, 1914, 1922) and others. Also, a comparative account of the genitalia, as a basis for the phylo genetic study of different insect orders, was employed by Walker (1919), Sharp and Muir (1912), Singh-Pruthi (1925) and Cole (1927), in Orthop tera, Coleoptera, Hemiptera and the Diptera respectively. It is sur prising, work of this nature having been found so useful in these groups, that an important order like the Lepidoptera should have escaped careful analysis at the hands of the morphologists. -
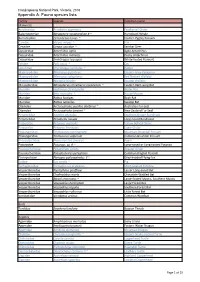
Report-VIC-Croajingolong National Park-Appendix A
Croajingolong National Park, Victoria, 2016 Appendix A: Fauna species lists Family Species Common name Mammals Acrobatidae Acrobates pygmaeus Feathertail Glider Balaenopteriae Megaptera novaeangliae # ~ Humpback Whale Burramyidae Cercartetus nanus ~ Eastern Pygmy Possum Canidae Vulpes vulpes ^ Fox Cervidae Cervus unicolor ^ Sambar Deer Dasyuridae Antechinus agilis Agile Antechinus Dasyuridae Antechinus mimetes Dusky Antechinus Dasyuridae Sminthopsis leucopus White-footed Dunnart Felidae Felis catus ^ Cat Leporidae Oryctolagus cuniculus ^ Rabbit Macropodidae Macropus giganteus Eastern Grey Kangaroo Macropodidae Macropus rufogriseus Red Necked Wallaby Macropodidae Wallabia bicolor Swamp Wallaby Miniopteridae Miniopterus schreibersii oceanensis ~ Eastern Bent-wing Bat Muridae Hydromys chrysogaster Water Rat Muridae Mus musculus ^ House Mouse Muridae Rattus fuscipes Bush Rat Muridae Rattus lutreolus Swamp Rat Otariidae Arctocephalus pusillus doriferus ~ Australian Fur-seal Otariidae Arctocephalus forsteri ~ New Zealand Fur Seal Peramelidae Isoodon obesulus Southern Brown Bandicoot Peramelidae Perameles nasuta Long-nosed Bandicoot Petauridae Petaurus australis Yellow Bellied Glider Petauridae Petaurus breviceps Sugar Glider Phalangeridae Trichosurus cunninghami Mountain Brushtail Possum Phalangeridae Trichosurus vulpecula Common Brushtail Possum Phascolarctidae Phascolarctos cinereus Koala Potoroidae Potorous sp. # ~ Long-nosed or Long-footed Potoroo Pseudocheiridae Petauroides volans Greater Glider Pseudocheiridae Pseudocheirus peregrinus -

Otter Slough Conservation Area (Stoddard County, Missouri) by Hugo L
SOUTHERN LEPIDOPTERISTS’ NEWS VOLUME 43 NO. 2 (2021), PG. 159 A LEPIDOPTERA BIODIVERSITY BLITZ AT THE OTTER SLOUGH CONSERVATION AREA (STODDARD COUNTY, MISSOURI) BY HUGO L. KONS JR. 1 & ROBERT J. BORTH 2 ABSTRACT We conducted a Lepidoptera biodiversity blitz on 3 and Catocala crataegi complex, representing the most 4 June 2018 at the Otter Slough Conservation Area in northerly locality that we are aware of for these Stoddard County, Missouri. We documented as many phenotypes. Recent material was needed for DNA Lepidoptera species as possible with MV/UV lights, sequencing. rotten banana/brown sugar bait, and diurnal collecting with nets. We present records for 235 species, including From 3-4 June 2018 we visited the Otter Slough 193 Macrolepidoptera and 19 Rhopalocera 3. Habitats Conservation Area to sample Catocala and document as sampled include hydric hardwood forest, cypress many other co-occurring Lepidoptera species as swamp, open wetlands, and field. Examples of some possible. This paper reports the Macrolepidoptera and species are shown on 15 color plates of live photos and Rhopalocera species recorded during this survey. This pinned specimens research was conducted under Wildlife Collectors Permit #17910 issued by the Missouri Department of INTRODUCTION Conservation. The Otter Slough Conservation Area is a 4,866 acre area MATERIALS AND METHODS including hydric hardwood forest (Figure 2:B, E-H), cypress-tupelo swamp (Figure 2:A), open marsh with Lepidoptera were sampled with a 400 watt MV cattails, sedge meadow, and cypress (Figure 2:D), illuminated sheet, 175 watt MV light trap, 15 watt UV mowed field (Figure 2:C (middle)), and slough habitats. -

Online Dictionary of Invertebrate Zoology: A
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Armand R. Maggenti Online Dictionary of Invertebrate Zoology Parasitology, Harold W. Manter Laboratory of September 2005 Online Dictionary of Invertebrate Zoology: A Mary Ann Basinger Maggenti University of California-Davis Armand R. Maggenti University of California, Davis Scott Lyell Gardner University of Nebraska - Lincoln, [email protected] Follow this and additional works at: https://digitalcommons.unl.edu/onlinedictinvertzoology Part of the Zoology Commons Maggenti, Mary Ann Basinger; Maggenti, Armand R.; and Gardner, Scott Lyell, "Online Dictionary of Invertebrate Zoology: A" (2005). Armand R. Maggenti Online Dictionary of Invertebrate Zoology. 16. https://digitalcommons.unl.edu/onlinedictinvertzoology/16 This Article is brought to you for free and open access by the Parasitology, Harold W. Manter Laboratory of at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Armand R. Maggenti Online Dictionary of Invertebrate Zoology by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Online Dictionary of Invertebrate Zoology 2 abdominal filament see cercus A abdominal ganglia (ARTHRO) Ganglia of the ventral nerve cord that innervate the abdomen, each giving off a pair of principal nerves to the muscles of the segment; located between the alimentary canal and the large ventral mus- cles. abactinal a. [L. ab, from; Gr. aktis, ray] (ECHINOD) Of or per- taining to the area of the body without tube feet that nor- abdominal process (ARTHRO: Crustacea) In Branchiopoda, mally does not include the madreporite; not situated on the fingerlike projections on the dorsal surface of the abdomen. ambulacral area; abambulacral. abactinally adv. abdominal somite (ARTHRO: Crustacea) Any single division of abambulacral see abactinal the body between the thorax and telson; a pleomere; a pleonite.