Raman Scattering Theory Introduction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Glossary Physics (I-Introduction)
1 Glossary Physics (I-introduction) - Efficiency: The percent of the work put into a machine that is converted into useful work output; = work done / energy used [-]. = eta In machines: The work output of any machine cannot exceed the work input (<=100%); in an ideal machine, where no energy is transformed into heat: work(input) = work(output), =100%. Energy: The property of a system that enables it to do work. Conservation o. E.: Energy cannot be created or destroyed; it may be transformed from one form into another, but the total amount of energy never changes. Equilibrium: The state of an object when not acted upon by a net force or net torque; an object in equilibrium may be at rest or moving at uniform velocity - not accelerating. Mechanical E.: The state of an object or system of objects for which any impressed forces cancels to zero and no acceleration occurs. Dynamic E.: Object is moving without experiencing acceleration. Static E.: Object is at rest.F Force: The influence that can cause an object to be accelerated or retarded; is always in the direction of the net force, hence a vector quantity; the four elementary forces are: Electromagnetic F.: Is an attraction or repulsion G, gravit. const.6.672E-11[Nm2/kg2] between electric charges: d, distance [m] 2 2 2 2 F = 1/(40) (q1q2/d ) [(CC/m )(Nm /C )] = [N] m,M, mass [kg] Gravitational F.: Is a mutual attraction between all masses: q, charge [As] [C] 2 2 2 2 F = GmM/d [Nm /kg kg 1/m ] = [N] 0, dielectric constant Strong F.: (nuclear force) Acts within the nuclei of atoms: 8.854E-12 [C2/Nm2] [F/m] 2 2 2 2 2 F = 1/(40) (e /d ) [(CC/m )(Nm /C )] = [N] , 3.14 [-] Weak F.: Manifests itself in special reactions among elementary e, 1.60210 E-19 [As] [C] particles, such as the reaction that occur in radioactive decay. -

DEVELOPMENT of a RAMAN SPECTROMETER to STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K
BARC/2011/E/003 BARC/2011/E/003 DEVELOPMENT OF A RAMAN SPECTROMETER TO STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K. Sarkar and Tulsi Mukherjee Radiation & Photochemistry Division 2011 BARC/2011/E/003 GOVERNMENT OF INDIA ATOMIC ENERGY COMMISSION BARC/2011/E/003 DEVELOPMENT OF A RAMAN SPECTROMETER TO STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K. Sarkar and Tulsi Mukherjee Radiation & Photochemistry Division BHABHA ATOMIC RESEARCH CENTRE MUMBAI, INDIA 2011 BARC/2011/E/003 BIBLIOGRAPHIC DESCRIPTION SHEET FOR TECHNICAL REPORT (as per IS : 9400 - 1980) 01 Security classification : Unclassified 02 Distribution : External 03 Report status : New 04 Series : BARC External 05 Report type : Technical Report 06 Report No. : BARC/2011/E/003 07 Part No. or Volume No. : 08 Contract No. : 10 Title and subtitle : Development of a Raman spectrometer to study surface-enhanced Raman scattering 11 Collation : 31 p., 15 figs. 13 Project No. : 20 Personal author(s) : Nandita Biswas; Ridhima Chadha; Sudhir Kapoor; Sisir K. Sarkar; Tulsi Mukherjee 21 Affiliation of author(s) : Radiation and Photochemistry Division , Bhabha Atomic Research Centre, Mumbai 22 Corporate author(s) : Bhabha Atomic Research Centre, Mumbai - 400 085 23 Originating unit : Radiation and Photochemistry Division, BARC, Mumbai 24 Sponsor(s) Name : Department of Atomic Energy Type : Government Contd... BARC/2011/E/003 30 Date of submission : January 2011 31 Publication/Issue date : February 2011 40 Publisher/Distributor : Head, Scientific Information Resource Division, Bhabha Atomic Research Centre, Mumbai 42 Form of distribution : Hard copy 50 Language of text : English 51 Language of summary : English, Hindi 52 No. -

12 Light Scattering AQ1
12 Light Scattering AQ1 Lev T. Perelman CONTENTS 12.1 Introduction ......................................................................................................................... 321 12.2 Basic Principles of Light Scattering ....................................................................................323 12.3 Light Scattering Spectroscopy ............................................................................................325 12.4 Early Cancer Detection with Light Scattering Spectroscopy .............................................326 12.5 Confocal Light Absorption and Scattering Spectroscopic Microscopy ............................. 329 12.6 Light Scattering Spectroscopy of Single Nanoparticles ..................................................... 333 12.7 Conclusions ......................................................................................................................... 335 Acknowledgment ........................................................................................................................... 335 References ...................................................................................................................................... 335 12.1 INTRODUCTION Light scattering in biological tissues originates from the tissue inhomogeneities such as cellular organelles, extracellular matrix, blood vessels, etc. This often translates into unique angular, polari- zation, and spectroscopic features of scattered light emerging from tissue and therefore information about tissue -

Solutions 7: Interacting Quantum Field Theory: Λφ4
QFT PS7 Solutions: Interacting Quantum Field Theory: λφ4 (4/1/19) 1 Solutions 7: Interacting Quantum Field Theory: λφ4 Matrix Elements vs. Green Functions. Physical quantities in QFT are derived from matrix elements M which represent probability amplitudes. Since particles are characterised by having a certain three momentum and a mass, and hence a specified energy, we specify such physical calculations in terms of such \on-shell" values i.e. four-momenta where p2 = m2. For instance the notation in momentum space for a ! scattering in scalar Yukawa theory uses four-momenta labels p1 and p2 flowing into the diagram for initial states, with q1 and q2 flowing out of the diagram for the final states, all of which are on-shell. However most manipulations in QFT, and in particular in this problem sheet, work with Green func- tions not matrix elements. Green functions are defined for arbitrary values of four momenta including unphysical off-shell values where p2 6= m2. So much of the information encoded in a Green function has no obvious physical meaning. Of course to extract the corresponding physical matrix element from the Greens function we would have to apply such physical constraints in order to get the physics of scattering of real physical particles. Alternatively our Green function may be part of an analysis of a much bigger diagram so it represents contributions from virtual particles to some more complicated physical process. ∗1. The full propagator in λφ4 theory Consider a theory of a real scalar field φ 1 1 λ L = (@ φ)(@µφ) − m2φ2 − φ4 (1) 2 µ 2 4! (i) A theory with a gφ3=(3!) interaction term will not have an energy which is bounded below. -
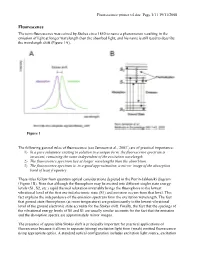
The Term Fluorescence Was Coined by Stokes Circa 1850 to Name A
Fluorescence primer v4.doc Page 1/11 19/11/2008 Fluorescence The term fluorescence was coined by Stokes circa 1850 to name a phenomenon resulting in the emission of light at longer wavelength than the absorbed light, and his name is still used to describe the wavelength shift (Figure 1A). Figure 1 The following general rules of fluorescence (see Jameson et al., 20031) are of practical importance: 1) In a pure substance existing in solution in a unique form, the fluorescence spectrum is invariant, remaining the same independent of the excitation wavelength. 2) The fluorescence spectrum lies at longer wavelengths than the absorbtion. 3) The fluorescence spectrum is, to a good approximation, a mirror image of the absorption band of least frequency These rules follow from quantum optical considerations depicted in the Perrin-Jablonski diagram (Figure 1B). Note that although the fluorophore may be excited into different singlet state energy levels (S1, S2, etc.) rapid thermal relaxation invariably brings the fluorophore to the lowest vibrational level of the first excited electronic state (S1) and emission occurs from that level. This fact explains the independence of the emission spectrum from the excitation wavelength. The fact that ground state fluorophores (at room temperature) are predominantly in the lowest vibrational level of the ground electronic state accounts for the Stokes shift. Finally, the fact that the spacings of the vibrational energy levels of S0 and S1 are usually similar accounts for the fact that the emission and the absorption spectra are approximately mirror images. The presence of appreciable Stokes shift is principally important for practical applications of fluorescence because it allows to separate (strong) excitation light from (weak) emitted fluorescence using appropriate optics. -

Resonant Raman Spectroscopy of Nanotubes
10.1098/rsta.2004.1444 Resonant Raman spectroscopy of nanotubes By Christian Thomsen1, Stephanie Reich2 and Janina Maultzsch1 1Technische Universit¨at Berlin, Hardenbergstraße 36, 10623 Berlin, Germany 2Department of Engineering, University of Cambridge, Trumpington Street, Cambridge CB2 1PZ, UK ([email protected]) Published online 28 September 2004 Single and double resonances in Raman scattering are introduced and six criteria for the observation and identification of double resonances stated. The experimental situation in carbon nanotubes is reviewed in view of these criteria. The evidence for the D mode and the high-energy mode is found to be overwhelming for a double- resonance process to take place, whereas the nature of the radial breathing-mode Raman process remains undecided at this point. Consequences for the application of Raman scattering to the characterization of nanotubes are discussed. Keywords: carbon nanotubes; double resonance; Raman scattering; defects 1. Introduction Raman scattering in carbon nanotubes has developed into a method of choice in the investigation of their physical properties and their characterization. The study of electronic resonances in the Raman spectra, a method which has been used exten- sively, for example, in work on semiconductors (Cardona 1982), gives us a wealth of information about the electronic band structure of a material. This is also true for the Raman work on carbon nanotubes, where resonance studies have moved into the focus of research. Traditional Raman studies of carbon nanotubes focus on the radial breathing mode (RBM), a mode where all atoms vibrate in phase in the radial direction (Dresselhaus et al. 1995). Its frequency, as can easily be shown (Jishi et al. -

Raman Scattering and Fluorescence
Fluorescence 01 Raman Scattering and Fluorescence Introduction The existence of such virtual states also explains why the non-resonance Raman effect Raman scattering and Fluorescence emission does not depend on the wavelength of the are two competing phenomena, which have excitation, since no real states are involved in similar origins. Generally, a laser photon this interaction mechanism. In fact, the Raman bounces off a molecule and looses a certain spectrum generally does not depend on the amount of energy that allows the molecule to laser excitation. vibrate (Stokes process). The scattered photon is therefore less energetic and the associated However, when the energy of the excitation light exhibits a frequency shift. The various photon gets close to the transition energy frequency shifts associated with different between two electronic states, one then deals molecular vibrations give rise to a spectrum, with resonance Raman or resonance that is characteristic of a specific compound. fluorescence (fig.1, case (d)). The basic difference between these two processes is In contrast, fluorescence or luminescence related to the time scales involved, as well as emission follows an absorption process. For a with the nature of the so-called intermediate better understanding, one can refer to the states. In contrast with resonant fluorescent, diagram below. relaxed fluorescence results from the emission of a photon from the lowest vibrational level of an excited electronic state, following a direct absorption of the photon and relaxation of the molecule from its vibrationally excited level of the electronic state back to the lowest vibrational level of the electronic state. A fluorescence process typically requires more than 10-9 s. -

The Basic Interactions Between Photons and Charged Particles With
Outline Chapter 6 The Basic Interactions between • Photon interactions Photons and Charged Particles – Photoelectric effect – Compton scattering with Matter – Pair productions Radiation Dosimetry I – Coherent scattering • Charged particle interactions – Stopping power and range Text: H.E Johns and J.R. Cunningham, The – Bremsstrahlung interaction th physics of radiology, 4 ed. – Bragg peak http://www.utoledo.edu/med/depts/radther Photon interactions Photoelectric effect • Collision between a photon and an • With energy deposition atom results in ejection of a bound – Photoelectric effect electron – Compton scattering • The photon disappears and is replaced by an electron ejected from the atom • No energy deposition in classical Thomson treatment with kinetic energy KE = hν − Eb – Pair production (above the threshold of 1.02 MeV) • Highest probability if the photon – Photo-nuclear interactions for higher energies energy is just above the binding energy (above 10 MeV) of the electron (absorption edge) • Additional energy may be deposited • Without energy deposition locally by Auger electrons and/or – Coherent scattering Photoelectric mass attenuation coefficients fluorescence photons of lead and soft tissue as a function of photon energy. K and L-absorption edges are shown for lead Thomson scattering Photoelectric effect (classical treatment) • Electron tends to be ejected • Elastic scattering of photon (EM wave) on free electron o at 90 for low energy • Electron is accelerated by EM wave and radiates a wave photons, and approaching • No -

Stokes Shift
Stokes shift ticular molecular structure. If a material has a direct bandgap in the range of visible light, the light shining on it is absorbed, causing electrons to become excited to a higher energy state. The electrons remain in the ex- cited state for about 10−8 seconds. This number varies over several orders of magnitude depending on the sam- ple, and is known as the fluorescence lifetime of the sam- ple. After losing a small amount of energy in some way (hence the longer wavelength), the molecule returns to the ground state and energy is emitted. 2 References Absorption and emission spectra of Rhodamine 6G with ~25 nm [1] Gispert, J.R. (2008). Coordination Chemistry. Wiley- Stokes shift VCH. p. 483. ISBN 3-527-31802-X. [2] Albani, J.R. (2004). Structure and Dynamics of Macro- Not to be confused with Stark shift. molecules: Absorption and Fluorescence Studies. Elsevier. p. 58. ISBN 0-444-51449-X. Stokes shift is the difference (in wavelength or frequency [3] Lakowicz, J.R. 1983. Principles of Fluorescence Spec- units) between positions of the band maxima of the troscopy, Plenum Press, New York. ISBN 0-387-31278- absorption and emission spectra (fluorescence and Raman 1. being two examples) of the same electronic transition.[1] [4] Guilbault, G.G. 1990. Practical Fluorescence, Second It is named after Irish physicist George G. Stokes.[2][3][4] Edition, Marcel Dekker, Inc., New York. ISBN 0-8247- When a system (be it a molecule or atom) absorbs a 8350-6. photon, it gains energy and enters an excited state. -

Fiber Amplifiers and Fiber Lasers Based on Stimulated Raman
micromachines Review Fiber Amplifiers and Fiber Lasers Based on Stimulated Raman Scattering: A Review Luigi Sirleto * and Maria Antonietta Ferrara National Research Council (CNR), Institute of Applied Sciences and Intelligent Systems, Via Pietro Castellino 111, 80131 Naples, Italy; [email protected] * Correspondence: [email protected] Received: 10 January 2020; Accepted: 24 February 2020; Published: 26 February 2020 Abstract: Nowadays, in fiber optic communications the growing demand in terms of transmission capacity has been fulfilling the entire spectral band of the erbium-doped fiber amplifiers (EDFAs). This dramatic increase in bandwidth rules out the use of EDFAs, leaving fiber Raman amplifiers (FRAs) as the key devices for future amplification requirements. On the other hand, in the field of high-power fiber lasers, a very attractive option is provided by fiber Raman lasers (FRLs), due to their high output power, high efficiency and broad gain bandwidth, covering almost the entire near-infrared region. This paper reviews the challenges, achievements and perspectives of both fiber Raman amplifier and fiber Raman laser. They are enabling technologies for implementation of high-capacity optical communication systems and for the realization of high power fiber lasers, respectively. Keywords: stimulated raman scattering; fiber optics; amplifiers; lasers; optical communication systems 1. Introduction Optical communication systems require optoelectronic devices, such as sources, detectors and so on, and utilize fiber optics to transmit the light carrying the signals impressed by modulators. Optical fibers are affected by chromatic dispersion, losses, and nonlinearity. Dispersion control is, usually, achieved via fiber geometry and material composition. Losses limit the transmission distance in modern long haul fiber-optic communication systems, so in order to boost a weak signal, optical amplifiers have been developed. -

Optical Light Manipulation and Imaging Through Scattering Media
Optical light manipulation and imaging through scattering media Thesis by Jian Xu In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy CALIFORNIA INSTITUTE OF TECHNOLOGY Pasadena, California 2020 Defended September 2, 2020 ii © 2020 Jian Xu ORCID: 0000-0002-4743-2471 All rights reserved except where otherwise noted iii ACKNOWLEDGEMENTS Foremost, I would like to express my sincere thanks to my advisor Prof. Changhuei Yang, for his constant support and guidance during my PhD journey. He created a research environment with a high degree of freedom and encouraged me to explore fields with a lot of unknowns. From his ethics of research, I learned how to be a great scientist and how to do independent research. I also benefited greatly from his high standards and rigorous attitude towards work, with which I will keep in my future path. My committee members, Professor Andrei Faraon, Professor Yanbei Chen, and Professor Palghat P. Vaidyanathan, were extremely supportive. I am grateful for the collaboration and research support from Professor Andrei Faraon and his group, the helpful discussions in optics and statistics with Professor Yanbei Chen, and the great courses and solid knowledge of signal processing from Professor Palghat P. Vaidyanathan. I would also like to thank the members from our lab. Dr. Haojiang Zhou introduced me to the wavefront shaping field and taught me a lot of background knowledge when I was totally unfamiliar with the field. Dr. Haowen Ruan shared many innovative ideas and designs with me, from which I could understand the research field from a unique perspective. -

Theoretical Investigation of Stokes Shifts and Reaction Pathways
Theoretical Investigation of Stokes Shifts and Reaction Pathways by Laken M. Top - ARI IVS Associate of Arts, Science Option Yakima Valley Community College, 2007 Bachelor of Science, Chemistry and Mathematics RES University of Idaho, 2009 SUBMITTED TO THE DEPARTMENT OF CHEMISTRY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE IN CHEMISTRY AT THE MASSACHUSETTS INSTITUTE OF TECHNOLOGY SEPTEMBER 2012 @ 2012 Massachusetts Institute of Technology. All rights reserved. 1 1-/ Signature of Author: Department of Chemistry July 12, 2012 Certified by: I Troy Van Voorhis ,1 Associate Professor of Chemistry Thesis Supervisor I / Certified by: f -y Jeffrey C. Grossman Associate Professor of Materials Science and Engineering Thesis Supervisor Accepted by: Robert W. Field Professor of Chemistry Chairman, Department Committee on Graduate Theses 1 Theoretical Investigation of Stokes Shifts and Reaction Pathways by Laken M. Top Submitted to the Department of Chemistry on July 12, 2012, in partial fulfillment of the requirements for the degree of Master of Science in Chemistry Abstract Solar thermal fuels and fluorescent solar concentrators provide two ways in which the energy from the sun can be harnessed and stored. While much progress has been made in recent years, there is still much more to learn about the way that these applications work and more efficient materials are needed to make this a feasible source of renewable energy. Theoretical chemistry is a powerful tool which can provide insight into the processes involved and the properties of materials, allowing us to predict substances that might improve the efficiency of these devices. In this work, we explore how the delta self-consistent field method performs for the calculation of Stokes shifts for conjugated dyes.