THE CELL BIOLOGICAL BASIS of CILIARY DISEASE • MARSHALL 19 and Channels Are Located in This Specialized Membrane, Which References Are Crucial for Ciliary Function
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Educational Paper Ciliopathies
Eur J Pediatr (2012) 171:1285–1300 DOI 10.1007/s00431-011-1553-z REVIEW Educational paper Ciliopathies Carsten Bergmann Received: 11 June 2011 /Accepted: 3 August 2011 /Published online: 7 September 2011 # The Author(s) 2011. This article is published with open access at Springerlink.com Abstract Cilia are antenna-like organelles found on the (NPHP) . Ivemark syndrome . Meckel syndrome (MKS) . surface of most cells. They transduce molecular signals Joubert syndrome (JBTS) . Bardet–Biedl syndrome (BBS) . and facilitate interactions between cells and their Alstrom syndrome . Short-rib polydactyly syndromes . environment. Ciliary dysfunction has been shown to Jeune syndrome (ATD) . Ellis-van Crefeld syndrome (EVC) . underlie a broad range of overlapping, clinically and Sensenbrenner syndrome . Primary ciliary dyskinesia genetically heterogeneous phenotypes, collectively (Kartagener syndrome) . von Hippel-Lindau (VHL) . termed ciliopathies. Literally, all organs can be affected. Tuberous sclerosis (TSC) . Oligogenic inheritance . Modifier. Frequent cilia-related manifestations are (poly)cystic Mutational load kidney disease, retinal degeneration, situs inversus, cardiac defects, polydactyly, other skeletal abnormalities, and defects of the central and peripheral nervous Introduction system, occurring either isolated or as part of syn- dromes. Characterization of ciliopathies and the decisive Defective cellular organelles such as mitochondria, perox- role of primary cilia in signal transduction and cell isomes, and lysosomes are well-known -

Renal Cystic Disorders Infosheet 6-14-19
Next Generation Sequencing Panel for Renal Cystic Disorders Clinical Features: Renal cystic diseases are a genetically heterogeneous group of conditions characterized By isolated renal disease or renal cysts in conjunction with extrarenal features (1). Age of onset of renal cystic disease ranges from neonatal to adult onset. Common features of renal cystic diseases include renal insufficiency and progression to end stage renal disease (ESRD). Identification of the genetic etiology of renal cystic disease can aid in appropriate clinical management of the affected patient. Our Renal Cystic Disorders Panel includes sequence and deletion/duplicaton analysis of all 79 genes listed below. Renal Cystic Disorders Sequencing Panel AHI1 BMPER HNF1B NEK8 TCTN3 WDPCP ANKS6 C5orf42 IFT27 NOTCH2 TFAP2A WDR19 ARL13B CC2D2A IFT140 NPHP1 TMEM107 XPNPEP3 ARL6 CDC73 IFT172 NPHP3 TMEM138 ZNF423 B9D1 CEP104 INPP5E NPHP4 TMEM216 B9D2 CEP120 INVS OFD1 TMEM231 BBIP1 CEP164 IQCB1 PDE6D TMEM237 BBS1 CEP290 JAG1 PKD2 TMEM67 BBS10 CEP41 KIAA0556 PKHD1 TRIM32 BBS12 CEP83 KIAA0586 REN TSC1 BBS2 CRB2 KIF14 RPGRIP1L TSC2 BBS4 CSPP1 KIF7 SALL1 TTC21B BBS5 DCDC2 LZTFL1 SDCCAG8 TTC8 BBS7 GLIS2 MKKS TCTN1 UMOD BBS9 GLIS3 MKS1 TCTN2 VHL Disorder Genes Inheritance Clinical features/molecular genetics Bardet Biedl ARL6 AR Bardet-Biedl syndrome (BBS) is an autosomal syndrome BBS1 recessive multi-systemic ciliopathy characterized By BBS10 retinal dystrophy, oBesity, postaxial polydactyly, BBS12 leaning difficulties, renal involvement and BBS2 genitourinary abnormalities (2). Visual prognosis is BBS4 poor, and the mean age of legal Blindness is 15.5 BBS5 years. Birth weight is typically normal But significant BBS7 weight gain Begins within the first year. Renal BBS9 disease is a major cause of morBidity and mortality. -

Novel Splicing Variant C. 208+2T>C in BBS5 Segregates with Bardet–Biedl
Bioscience Reports (2019) 39 BSR20181544 https://doi.org/10.1042/BSR20181544 Research Article Novel splicing variant c. 208+2T>CinBBS5 segregates with Bardet–Biedl syndrome in an Iranian family by targeted exome sequencing Saber Imani1,*, Jingliang Cheng1,*,JiewenFu1,2,*, Abdolkarim Mobasher-Jannat3,*, Chunli Wei1,4, Saman Mohazzab-Torabi5, Khosrow Jadidi6, Mohammad Hossein Khosravi3, Marzieh Dehghan Shasaltaneh7, Lisha Yang1, Md. Asaduzzaman Khan1 and Junjiang Fu1,4 1Key Laboratory of Epigenetics and Oncology, Research Center for Preclinical Medicine, Southwest Medical University, Luzhou, Sichuan, China; 2Institute of Medical Technology, Xiangtan Medicine and Health Vocational College, Xiangtan, Hunan, China; 3Student Research Committee, Baqiyatallah University of Medical Sciences, Tehran, Iran; 4State Key Laboratory of Quality Research in Chinese Medicine, Macau Institute for Applied Research in Medicine and Health, Macau University of Science and Technology, Macau (SAR), China; 5Noor Ophthalmology Research Center, Noor Eye Hospital, Tehran, Iran; 6Department of Ophthalmology, Bina Eye Hospital Research Center, Tehran, Iran; 7Department of Biology, Faculty of Science, University of Zanjan, Zanjan, Iran Correspondence: Junjiang Fu ([email protected]) Bardet–Biedl syndrome (BBS) is a rare genetically heterogeneous ciliopathy which accom- panies retinitis pigmentosa (RP). However, the BBS5 mutation remains unclear in Iranians with BBS. The purpose of study is to evaluate genetic analyses of a BBS Iranian family us- ing targetted exome sequencing (TES). A male 11-year-old proband and three related fam- ily members were recruited. Biochemical tests, electrocardiography and visual acuity test- ing, such as funduscopic, fundus photography (FP), optical coherence tomography (OCT), and standard electroretinography, were conducted. Molecular analysis and high-throughput DNA sequence analysis were performed. -

Ciliopathies
T h e new england journal o f medicine Review article Mechanisms of Disease Robert S. Schwartz, M.D., Editor Ciliopathies Friedhelm Hildebrandt, M.D., Thomas Benzing, M.D., and Nicholas Katsanis, Ph.D. iverse developmental and degenerative single-gene disor- From the Howard Hughes Medical Insti- ders such as polycystic kidney disease, nephronophthisis, retinitis pigmen- tute and the Departments of Pediatrics and Human Genetics, University of Michi- tosa, the Bardet–Biedl syndrome, the Joubert syndrome, and the Meckel gan Health System, Ann Arbor (F.H.); the D Renal Division, Department of Medicine, syndrome may be categorized as ciliopathies — a recent concept that describes dis- eases characterized by dysfunction of a hairlike cellular organelle called the cilium. Center for Molecular Medicine, and Co- logne Cluster of Excellence in Cellular Most of the proteins that are altered in these single-gene disorders function at the Stress Responses in Aging-Associated Dis- level of the cilium–centrosome complex, which represents nature’s universal system eases, University of Cologne, Cologne, for cellular detection and management of external signals. Cilia are microtubule- Germany (T.B.); and the Center for Hu- man Disease Modeling and the Depart- based structures found on almost all vertebrate cells. They originate from a basal ments of Pediatrics and Cell Biology, body, a modified centrosome, which is the organelle that forms the spindle poles Duke University Medical Center, Durham, during mitosis. The important role that the cilium–centrosome complex plays in NC (N.K.). Address reprint requests to Dr. Hildebrandt at Howard Hughes Med- the normal function of most tissues appears to account for the involvement of mul- ical Institute, Departments of Pediatrics tiple organ systems in ciliopathies. -

<Em>RPGRIP1</Em> and Cone-Rod Dystrophy in Dogs
University of Pennsylvania ScholarlyCommons Departmental Papers (Vet) School of Veterinary Medicine 2012 RPGRIP1 and Cone-Rod Dystrophy in Dogs Tatyana N. Kuznetsova Barbara Zangerl University of Pennsylvania, [email protected] Gustavo D. Aguirre University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/vet_papers Part of the Eye Diseases Commons, Geriatrics Commons, Medical Biotechnology Commons, Medical Genetics Commons, Medical Immunology Commons, Ophthalmology Commons, and the Veterinary Medicine Commons Recommended Citation Kuznetsova, T. N., Zangerl, B., & Aguirre, G. D. (2012). RPGRIP1 and Cone-Rod Dystrophy in Dogs. Retinal Degenerative Diseases: Advances in Experimental Medicine and Biology, 723 321-328. http://dx.doi.org/10.1007/978-1-4614-0631-0_42 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/vet_papers/85 For more information, please contact [email protected]. RPGRIP1 and Cone-Rod Dystrophy in Dogs Abstract Cone–rod dystrophies (crd) represent a group of progressive inherited blinding diseases characterized by primary dysfunction and loss of cone photoreceptors accompanying or preceding rod death. Recessive crd type 1 was described in dogs associated with an RPGRIP1 exon 2 mutation, but with lack of complete concordance between genotype and phenotype. This review highlights role of the RPGRIP1, a component of complex protein networks, and its function in the primary cilium, and discusses the potential mechanisms of genotype–phenotype discordance observed in dogs with the RPGRIP1 mutation. Keywords RPGRIP1, polymorphism, cone-rod dystrophy, protein network, photoreceptor cilia Disciplines Eye Diseases | Geriatrics | Medical Biotechnology | Medical Genetics | Medical Immunology | Ophthalmology | Veterinary Medicine This journal article is available at ScholarlyCommons: https://repository.upenn.edu/vet_papers/85 Published in final edited form as: Adv Exp Med Biol. -
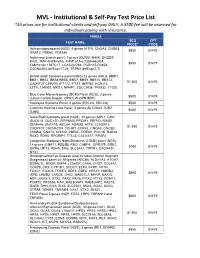
MVL - Institutional & Self-Pay Test Price List *All Prices Are for Institutional Clients and Self-Pay ONLY
MVL - Institutional & Self-Pay Test Price List *All prices are for institutional clients and self-pay ONLY. A $500 fee will be assessed for individuals paying with insurance. PANELS SEQ CPT TEST NAME PRICE* CODE Achromatopsiapanel (NGS), 6 genes (ATF6, CNGA3, CNGB3, $950 81479 GNAT2, PDE6C, PDE6H) Ashkenazi Jewish panel, 7 genes (CLRN1-N48K, DHDDS- K42E, MAK-K429insAlu, FAM161A-c.1355-6delCA, $550 81407 FAM161Ac.1567C>T, LCA5-Q279X, PCDH15-R245X, CACNA2D4-delExon17-26, TRPM1-delExon2-7) Bardet-Biedl Syndrome panel (NGS) 23 genes (ARL6, BBIP1, BBS1, BBS2, BBS4,BBS5, BBS7, BBS9, BBS10, BBS12, $1,500 81479 C8ORF37,CEP290, IFT172, IFT27, INPP5E, KCNJ13, LZTFL1,MKKS, MKS1, NPHP1, SDCCAG8, TRIM32, TTC8) Blue Cone Monochromacy (BCM) Panel (NGS), 2 genes $500 81479 (Locus Control Region, OPN1LW-OPN1MW) Cockayne Sydrome Panel, 2 genes (ERCC6, ERCC8) $500 81479 Common Hearing Loss Panel, 3 genes (SLC26A4, GJB2, $650 81479 GJB6) Cone-Rod Dystrophy panel (NGS), 33 genes (AIPL1, CRX, GUCA1A, GUCY2D, PITPNM3, PROM1, PRPH2, RIMS1, SEMA4A, UNC119, ABCA4, ADAM9, ATF6, C21ORF2, $1,950 81479 C8ORF37,CACNA2D4, CDHR1, CERKL, CNGA3, CNGB3, CNNM4, GNAT2, KCNV2, PDE6C, PDE6H, POC1B, RAB28, RAX2, RDH5, RPGRIP1, TTLL5, CACNA1F, RPGR) Congenital Stationary Night Blindness (CSNB) panel (NGS), 14 genes (GNAT1, PDE6B, RHO, CABP4, GPR179, GRK1, $950 81479 GRM6,LRIT3, RDH5, SAG, SLC24A1, TRPM1, CACNA1F, NYX) Developmental Eye Disease (also includes Anterior Segment Dysgenesis) panel v4: 59 genes (ABCB6, ALDH1A3, ATOH7, B3GALTL, BCOR, BMP4, c12orf57, CASK, CHD7, COL4A1, -

Glomerulocystic Kidney Disease in Mice with a Targeted Inactivation of Wwtr1
Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1 Zakir Hossain*†, Safiah Mohamed Ali*, Hui Ling Ko*, Jianliang Xu*, Chee Peng Ng‡, Ke Guo§, Zeng Qi§, Sathivel Ponniah*, Wanjin Hong‡, and Walter Hunziker*¶ *Epithelial Cell Biology Laboratory, ‡Membrane Biology Laboratory, and §Histology Unit, Institute of Molecular and Cell Biology, 61 Biopolis Drive, Republic of Singapore 138673 Edited by Christine E. Seidman, Harvard Medical School, Boston, MA, and approved November 29, 2006 (received for review June 22, 2006) Wwtr1 is a widely expressed 14-3-3-binding protein that regulates An increasing number of genes linked to PKD have been the activity of several transcription factors involved in develop- shown to encode proteins associated with the structure and/or ment and disease. To elucidate the physiological role of Wwtr1, we function of primary cilia, strongly suggesting that defects in the .(generated Wwtr1؊/؊ mice by homologous recombination. Surpris- ciliary apparatus play a central role in the etiology of PKD (4–6 ingly, although Wwtr1 is known to regulate the activity of Cbfa1, Primary or nonmotile cilia are structurally related to motile a transcription factor important for bone development, Wwtr1؊/؊ flagella of sperm and protozoa but serve as mechano- or mice show only minor skeletal defects. However, Wwtr1؊/؊ ani- chemosensors (reviewed in ref. 4). Cilia are anchored to the basal mals present with renal cysts that lead to end-stage renal disease. body, and their structural unit, the axoneme, consists of micro- Cysts predominantly originate from the dilation of Bowman’s tubules, dynein arms, and radial spoke proteins. Cilia assembly spaces and atrophy of glomerular tufts, reminiscent of glomeru- and maintenance involves the antero- and retrograde transport locystic kidney disease in humans. -

Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss
cells Review Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss 1, 1, 1 1,2,3 1 Gayle B. Collin y, Navdeep Gogna y, Bo Chang , Nattaya Damkham , Jai Pinkney , Lillian F. Hyde 1, Lisa Stone 1 , Jürgen K. Naggert 1 , Patsy M. Nishina 1,* and Mark P. Krebs 1,* 1 The Jackson Laboratory, Bar Harbor, Maine, ME 04609, USA; [email protected] (G.B.C.); [email protected] (N.G.); [email protected] (B.C.); [email protected] (N.D.); [email protected] (J.P.); [email protected] (L.F.H.); [email protected] (L.S.); [email protected] (J.K.N.) 2 Department of Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand 3 Siriraj Center of Excellence for Stem Cell Research, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand * Correspondence: [email protected] (P.M.N.); [email protected] (M.P.K.); Tel.: +1-207-2886-383 (P.M.N.); +1-207-2886-000 (M.P.K.) These authors contributed equally to this work. y Received: 29 February 2020; Accepted: 7 April 2020; Published: 10 April 2020 Abstract: Inherited retinal degeneration (RD) leads to the impairment or loss of vision in millions of individuals worldwide, most frequently due to the loss of photoreceptor (PR) cells. Animal models, particularly the laboratory mouse, have been used to understand the pathogenic mechanisms that underlie PR cell loss and to explore therapies that may prevent, delay, or reverse RD. Here, we reviewed entries in the Mouse Genome Informatics and PubMed databases to compile a comprehensive list of monogenic mouse models in which PR cell loss is demonstrated. -

NPHP1 Gene Deletion Is a Rare Cause of Joubert Syndrome Related Disorders
1of4 ELECTRONIC LETTER J Med Genet: first published as 10.1136/jmg.2004.027375 on 2 February 2005. Downloaded from NPHP1 gene deletion is a rare cause of Joubert syndrome related disorders M Castori, E M Valente, M A Donati, S Salvi, E Fazzi, E Procopio, T Galluccio, F Emma, B Dallapiccola, E Bertini, the Italian MTS Study Group* ............................................................................................................................... J Med Genet 2005;42:e9 (http://www.jmedgenet.com/cgi/content/full/42/2/e9). doi: 10.1136/jmg.2004.027375 oubert syndrome (JS) is an autosomal recessive disorder presenting with congenital hypotonia evolving into ataxia, Key points Jdevelopmental delay, and either oculomotor apraxia or abnormalities of respiratory pattern or both. JS is charac- N Joubert syndrome (JS) is a neurological disorder terised, using magnetic resonance imaging (MRI), by characterised by a complex cerebellar and brainstem cerebellar vermian hypoplasia and a complex brain stem malformation, the so called ‘‘molar tooth sign’’ (MTS). malformation called the ‘‘molar tooth sign’’ (MTS), consist- JS can be associated with several abnormalities in ing of thickened, elongated, and reoriented superior cerebel- other organs, identifying a large spectrum of ‘‘Joubert 1 lar peduncles and a deep interpeduncular fossa. JS has been syndrome related disorders’’ (JSRD). Isolated nephro- classified into two groups, A and B, the latter being nophthisis (NPH) is an autosomal recessive tubulointer- characterised by the occurrence of retinal and/or renal stitial medullary cystic kidney disease, which can be involvement.2 found in some JSRD. Among the four genes responsible Key associated features of JS are retinal dystrophy and for isolated NPH (NPHP1–4), NPHP1 deletions have nephronophthisis, but other manifestations include ocular been found in two families with JS plus NPH. -

1 AHI1 Mutations Cause Both Retinal
JMG Online First, published on September 9, 2005 as 10.1136/jmg.2005.036608 1 J Med Genet: first published as 10.1136/jmg.2005.036608 on 9 September 2005. Downloaded from AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome Melissa A. Parisi1, Daniel Doherty1, Melissa L. Eckert1, Dennis W.W. Shaw2, Hamit Ozyurek3, Sabiha Aysun3, Ozlem Giray4, Abdulrahman Al Swaid5, Saad Al Shahwan5, Noura Dohayan5, Eman Bakhsh6, Olafur S. Indridason7, William B. Dobyns8, Craig L. Bennett1, Phillip F. Chance1,9, Ian A. Glass1 Departments of 1Pediatrics, 2Radiology, and 9Neurology, Children’s Hospital and Regional Medical Center and the University of Washington, Seattle, WA, USA 3Department of Pediatrics, Hacettepe University, Ankara, Turkey 4Department of Pediatric Genetics, Dokuz Eylul University, Izmir, Turkey Departments of 5Paediatrics and 6Radiology, Riyadh Armed Forces Hospital, Riyadh, Saudi Arabia 7Department of Medicine, Landspitali-University Hospital, Reykjavik, Iceland 8Department of Human Genetics, University of Chicago, Chicago, IL, USA Corresponding Author: Melissa A. Parisi, MD, PhD, Division of Genetics and Developmental Medicine, Department of Pediatrics, University of Washington School of Medicine, Box 356320, 1959 NE Pacific St., Seattle, WA 98195, USA. ph: 206-987-2689; fax: 206-987-2495; email: [email protected] http://jmg.bmj.com/ on September 27, 2021 by guest. Protected copyright. Copyright Article author (or their employer) 2005. Produced by BMJ Publishing Group Ltd under licence. 2 J Med Genet: first published as 10.1136/jmg.2005.036608 on 9 September 2005. Downloaded from ABSTRACT Background: Joubert syndrome (JS) is an autosomal recessive disorder characterized by hypotonia, ataxia, mental retardation, altered respiratory pattern, abnormal eye movements, and a brain malformation known as the "molar tooth sign" (MTS) on cranial MRI. -

Cone Dystrophy Associated with a Novel Variant in the Terminal Codon of the RPGR-ORF15
G C A T T A C G G C A T genes Article Cone Dystrophy Associated with a Novel Variant in the Terminal Codon of the RPGR-ORF15 Vlasta Hadalin 1, Maja Šuštar 1, Marija Volk 2, Aleš Maver 2, Jana Sajovic 1, Martina Jarc-Vidmar 1, Borut Peterlin 2, Marko Hawlina 1 and Ana Fakin 1,* 1 Eye Hospital, University Medical Centre Ljubljana, Grabloviˇceva46, 1000 Ljubljana, Slovenia; [email protected] (V.H.); [email protected] (M.Š.); [email protected] (J.S.); [email protected] (M.J.-V.); [email protected] (M.H.) 2 Clinical Institute of Medical Genetics, University Medical Centre Ljubljana, Šlajmerjeva ulica 4, 1000 Ljubljana, Slovenia; [email protected] (M.V.); [email protected] (A.M.); [email protected] (B.P.) * Correspondence: [email protected] Abstract: Mutations in RPGRORF15 are associated with rod-cone or cone/cone-rod dystrophy, the latter associated with mutations at the distal end. We describe the phenotype associated with a novel variant in the terminal codon of the RPGRORF15 c.3457T>A (Ter1153Lysext*38), which results in a C-terminal extension. Three male patients from two families were recruited, aged 31, 35, and 38 years. Genetic testing was performed by whole exome sequencing. Filtered variants were analysed according to the population frequency, ClinVar database, the variant’s putative impact, and predicted pathogenicity; and were classified according to the ACMG guidelines. Examination included visual acuity (Snellen), colour vision (Ishihara), visual field, fundus autofluorescence (FAF), optical coherence tomography (OCT), and electrophysiology. All patients were myopic, and had Citation: Hadalin, V.; Šuštar, M.; central scotoma and reduced colour vision. -

HHS Public Access Author Manuscript
HHS Public Access Author manuscript Author Manuscript Author ManuscriptNature. Author ManuscriptAuthor manuscript; Author Manuscript available in PMC 2015 November 28. Published in final edited form as: Nature. 2015 May 28; 521(7553): 520–524. doi:10.1038/nature14269. Global genetic analysis in mice unveils central role for cilia in congenital heart disease You Li1,8, Nikolai T. Klena1,8, George C Gabriel1,8, Xiaoqin Liu1,7, Andrew J. Kim1, Kristi Lemke1, Yu Chen1, Bishwanath Chatterjee1, William Devine2, Rama Rao Damerla1, Chien- fu Chang1, Hisato Yagi1, Jovenal T. San Agustin5, Mohamed Thahir3,4, Shane Anderton1, Caroline Lawhead1, Anita Vescovi1, Herbert Pratt5, Judy Morgan6, Leslie Haynes6, Cynthia L. Smith6, Janan T. Eppig6, Laura Reinholdt6, Richard Francis1, Linda Leatherbury7, Madhavi K. Ganapathiraju3,4, Kimimasa Tobita1, Gregory J. Pazour5, and Cecilia W. Lo1,9 1Department of Developmental Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA 2Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA 3Department of Biomedical Informatics, University of Pittsburgh School of Medicine, Pittsburgh, PA 4Intelligent Systems Program, School of Arts and Sciences, University of Pittsburgh, Pittsburgh, PA 9Corresponding author. [email protected] Phone: 412-692-9901, Mailing address: Dept of Developmental Biology, Rangos Research Center, 530 45th St, Pittsburgh, PA, 15201. 8Co-first authors Author Contributions: Study design: CWL ENU mutagenesis, line cryopreservation, JAX strain datasheet construction: