First Draft Genome of the Sable, Martes Zibellina
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport Characteristics of Salt Ions in Soil Columns Planted with Tamarix Chinensis Under Different Groundwater Levels
RESEARCH ARTICLE Transport characteristics of salt ions in soil columns planted with Tamarix chinensis under different groundwater levels 1,2 1 2 1,2 1 1 Ximei Zhao , Jiangbao XiaID *, Weifeng Chen , Yinping Chen , Ying Fang , Fanzhu Qu 1 Shandong Key Laboratory of Eco-Environmental Science for the Yellow River Delta, Binzhou University, Binzhou, China, 2 College of Resources and Environment, Shandong Agriculture University, Tai'an, China * [email protected] a1111111111 a1111111111 a1111111111 a1111111111 Abstract a1111111111 The groundwater level is the main factor affecting the distribution of soil salinity and vegetation in the Yellow River Delta (YRD), China, but the response relationship between the spatial dis- tribution of soil salt ions and the groundwater level in the soil-Tamarix chinensis system remains unclear. In order to investigate the patterns of soil salt ions responding to groundwater OPEN ACCESS levels, in the `groundwater-soil-T. chinensis' system. Soil columns planted with T. chinensis, a Citation: Zhao X, Xia J, Chen W, Chen Y, Fang Y, constructive species in the YRD, were taken as the study object, and six groundwater levels Qu F (2019) Transport characteristics of salt ions in soil columns planted with Tamarix chinensis under (0.3, 0.6, 0.9, 1.2, 1.5 and 1.8 m) were simulated under saline mineralization. The results dem- + - different groundwater levels. PLoS ONE 14(4): onstrated the following: As affected by groundwater, Na and Cl were the main ions in the e0215138. https://doi.org/10.1371/journal. T. chinensis-planted soil column, with a trend of decreasing first and then increasing by the pone.0215138 + - increase of soil depth. -

A Complete Collection of Chinese Institutes and Universities For
Study in China——All China Universities All China Universities 2019.12 Please download WeChat app and follow our official account (scan QR code below or add WeChat ID: A15810086985), to start your application journey. Study in China——All China Universities Anhui 安徽 【www.studyinanhui.com】 1. Anhui University 安徽大学 http://ahu.admissions.cn 2. University of Science and Technology of China 中国科学技术大学 http://ustc.admissions.cn 3. Hefei University of Technology 合肥工业大学 http://hfut.admissions.cn 4. Anhui University of Technology 安徽工业大学 http://ahut.admissions.cn 5. Anhui University of Science and Technology 安徽理工大学 http://aust.admissions.cn 6. Anhui Engineering University 安徽工程大学 http://ahpu.admissions.cn 7. Anhui Agricultural University 安徽农业大学 http://ahau.admissions.cn 8. Anhui Medical University 安徽医科大学 http://ahmu.admissions.cn 9. Bengbu Medical College 蚌埠医学院 http://bbmc.admissions.cn 10. Wannan Medical College 皖南医学院 http://wnmc.admissions.cn 11. Anhui University of Chinese Medicine 安徽中医药大学 http://ahtcm.admissions.cn 12. Anhui Normal University 安徽师范大学 http://ahnu.admissions.cn 13. Fuyang Normal University 阜阳师范大学 http://fynu.admissions.cn 14. Anqing Teachers College 安庆师范大学 http://aqtc.admissions.cn 15. Huaibei Normal University 淮北师范大学 http://chnu.admissions.cn Please download WeChat app and follow our official account (scan QR code below or add WeChat ID: A15810086985), to start your application journey. Study in China——All China Universities 16. Huangshan University 黄山学院 http://hsu.admissions.cn 17. Western Anhui University 皖西学院 http://wxc.admissions.cn 18. Chuzhou University 滁州学院 http://chzu.admissions.cn 19. Anhui University of Finance & Economics 安徽财经大学 http://aufe.admissions.cn 20. Suzhou University 宿州学院 http://ahszu.admissions.cn 21. -

Partner Universities and Institutions List 1/ 19
Partner Universities and Institutions List (May 19, 2017) Total : 63 Countries, 506iversities & Institutions Signature Country University / Institution Agreement Level Date Afghanistan Kabul University Department Level 2014.01. Australia Curtin University University Level 2012.06. Australia Macquarie University University Level 2011.04. Australia Murdoch University University Level 2008.05. Australia Swinburne University of Technology College Level 2012.09. Australia The University of Adelaide University Level 2012.07. Australia University of New England University Level 2008.06. Australia University of the Sunshine Coast University Level 2008.01. Austria International Institute for Applied Systems Analysis (IIASA) Research Center Level 2014.07. Austria University of Applied Sciences Technikum Wien (FH Technikum Wien) University Level 2008.05. Azerbaijan Azerbaijan University of Architecture and Construction University Level 2012.01. University Level (Glocal Azerbaijan Baku State University 2007.02. Campus) Belarus Belarusian State University of Culture and Arts University Level 2007.05. Belgium Ghent University, Global Campus University Level 2016.03. Belgium Vesalius College University Level 2009.11. Brazil Mackenzie Presbyterian University University Level 2012.11. Brazil Universidade de Ribeirão Preto (UNAERP) University Level 2014.02. Brazil Universidade Estadual Paulista (UNESP) University Level 2013.05. Brazil Universidade Federal Fluminese University Level 2013.04. University Level (Glocal Cambodia Build Bright University 2012.07. Campus) University Level (Glocal Cambodia Pannasastra University 2012.03. Campus) Cambodia Royal University of Phnom Penh University Level 2011.01. Canada Centennial College University Level 2015.02. Canada Douglas College University Level 2008.03. Canada Lethbridge College University Level 2012.03. Canada McGill University Department Level 2013.12. 1/ 19 Partner Universities and Institutions List Signature Country University / Institution Agreement Level Date Graduate School Level 2007.02. -

An Inequality for Generalized Complete Elliptic Integral Li Yin1*, Li-Guo Huang1,Yong-Liwang2 and Xiu-Li Lin3
Yin et al. Journal of Inequalities and Applications (2017)2017:303 DOI 10.1186/s13660-017-1578-6 R E S E A R C H Open Access An inequality for generalized complete elliptic integral Li Yin1*, Li-Guo Huang1,Yong-LiWang2 and Xiu-Li Lin3 *Correspondence: [email protected] Abstract 1Department of Mathematics, Binzhou University, Binzhou City, In this paper, we show an elegant inequality involving the ratio of generalized Shandong Province 256603, China complete elliptic integrals of the first kind and generalize an interesting result of Alzer. Full list of author information is available at the end of the article MSC: 33E05 Keywords: generalized complete elliptic integrals; psi function; hypergeometric function; inequality 1 Introduction The generalized complete elliptic integral of the first kind is defined for r ∈ (0, 1) by πp 2 dθ 1 dt Kp(r)= 1 = 1 1 , 0 p p 1– p 0 p p p p 1– p (1 – r sinp θ) (1 – t ) (1 – r t ) where sinp θ is the generalized trigonometric function and 1 dt 2 1 1 πp =2 1 = B ,1– . 0 (1 – tp) p p p p The function sinp θ and the number πp play important roles in expressing the solu- tions of inhomogeneous eigenvalue problem of p-Laplacian –(|u|p–2u) = λ|u|p–2u with a boundary condition. These functions have some applications in the quasi-conformal the- ory, geometric function theory and the theory of Ramanujan modular equation. Báricz [1] established some Turán type inequalities for a Gauss hypergeometric function and for a generalized complete elliptic integral and showed a sharp bound for the generalized com- plete elliptic integral of the first kind in 2007. -

University of Leeds Chinese Accepted Institution List 2021
University of Leeds Chinese accepted Institution List 2021 This list applies to courses in: All Engineering and Computing courses School of Mathematics School of Education School of Politics and International Studies School of Sociology and Social Policy GPA Requirements 2:1 = 75-85% 2:2 = 70-80% Please visit https://courses.leeds.ac.uk to find out which courses require a 2:1 and a 2:2. Please note: This document is to be used as a guide only. Final decisions will be made by the University of Leeds admissions teams. -

2021 ICM Contest
2021 Interdisciplinary Contest in Modeling® Press Release—April 23, 2021 COMAP is pleased to announce the results of the 23nd annual influence between artists, and to identify revolutionaries. The E Interdisciplinary Contest in Modeling (ICM). This year 16,059 teams Problem asked teams to create a more equitable and sustainable food representing institutions from sixteen countries/regions participated in system. Students were also required to consider the timeline of the contest. Nineteen teams were designated as OUTSTANDING implementation and the obstacles to change for a region. The F representing the following schools: Problem considered the future of higher education by asking students to create a model to measure the health and sustainability of a national Beijing Normal University, China system of higher education. This problem required actionable policies Northwestern Polytechnical University, China to move a country to a healthier and more sustainable system based on Fudan University, China (AMS Award) the components they chose to include in their model and the country Shenzhen University, China (AMS Award) being considered. For all three problems, teams used pertinent data and South China University of Technology, China grappled with how phenomena internal and external to the system Shanghai Jiao Tong University, China (2) needed to be considered and measured. The student teams produced (INFORMS Award 2103649, INFORMS Award 2106028) creative and relevant solutions to these complex questions and built China University of Petroleum (East China), China models to handle the tasks assigned in the problems. The problems (Vilfredo Pareto Award) also required data analysis, creative modeling, and scientific Xidian University, China methodology, along with effective writing and visualization to Renmin University, China (SIAM Award) communicate their teams' results in a 25-page report. -
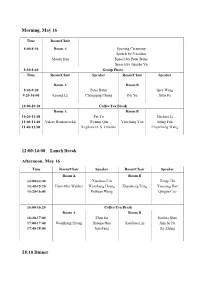
Program and Abstract of Talks
Morning, May 16 Time Room/Chair 8:00-8:30 Room A Opening Ceremony Speech by President Maoan Han Speech by Peter Bates Speech by Jianshe Yu 8:30-8:40 Group Photo Time Room/Chair Speaker Room/Chair Speaker Room A Room B 8:40-9:20 Peter Bates Qiru Wang 9:20-10:00 Kening Lu Chongqing Cheng Pei Yu Xilin Fu 10:00-10:20 Coffee/Tea Break Room A Room B 10:20-11:00 Pei Yu Michael Li 11:00-11:40 Valery Romanovskii Wenxin Qin Yuncheng You Meng Fan 11:40-12:00 Regilene D. S. Oliveira Chuncheng Wang 12:00-14:00 Lunch Break Afternoon, May 16 Time Room/Chair Speaker Room/Chair Speaker Room A Room B 14:00-14:40 Xiaobiao Lin Zengji Du 14:40-15:20 Hans-Otto Walther Wenzhang Huang Zhaosheng Feng Xiaoying Han 15:20-16:00 Peixuan Weng Qingjie Cao 16:00-16:20 Coffee/Tea Break Room A Room B 16:20-17:00 Zhen Jin Jianhua Shen 17:00-17:40 Wenzhang Huang Xiaojie Hou Xiaobiao Lin Xinchu Fu 17:40-18:00 Jian Fang Xu Zhang 18:10 Dinner Morning, May 17 Time Room/Chair Speaker Room/Chair Speaker Room A Room B 8:00-8:40 Kening Lu Zhaosheng Feng 8:40-9:20 Peter Bates Dongmei Xiao Jia Li Wei Ding 9:20-10:00 Huyi Hu Xiaosong Yang 10:00-10:20 Coffee/Tea Break Room A Room B 10:20-11:00 Yongluo Cao Yi Wang 11:00-11:40 Huyi Hu Wen Huang Daoyi Xu Fengjuan Chen 11:40-12:00 Yepeng Xing Kanat Shakenov 12:00-14:00 Lunch Break Afternoon, May 17 Time Room/Chair Speaker Room/Chair Speaker Room A Room B 14:00-14:40 Jia Li Valery Romanovskii 14:40-15:20 Huaiping Zhu Jifa Jiang Dongmei Xiao Xiang Zhang 15:20-16:00 Lin Wang Yuming Shi 16:00-16:20 Coffee/Tea Break Room A Room B 16:20-17:00 -

Theoretical and Experimental Study on the Degradation Mechanism of Atrazine in Fenton Cite This: RSC Adv.,2017,7,1581 Oxidation Treatment†
RSC Advances View Article Online PAPER View Journal | View Issue Theoretical and experimental study on the degradation mechanism of atrazine in Fenton Cite this: RSC Adv.,2017,7,1581 oxidation treatment† Xue Zhao,a Chenxi Zhang,b Shuguang Wang,*c Chao Songc and Xiang Li*d The residues of atrazine in surface and ground water will cause harm to human health as they are slowly biodegraded microbiologically. In this work, density functional theory (DFT) and the polarizable continuum model (PCM) were used to investigate the degradation of atrazine in an aqueous medium by Fenton oxidation technology. The results show that H atom abstraction pathways are more probable than both OH radical addition and Cl atom substitution pathways. Moreover, the H atom abstraction from the –CH– of –CH(CH3)2 group and –CH2– of –CH2CH3 group are expected to occur more easily. New dealkylation and alkyl oxidation mechanisms are proposed, in which water can act as a catalyst to Received 17th November 2016 reduce the reaction barrier dramatically. The stable intermediates and products: CH COCH , DEDIA, DIA, Accepted 17th December 2016 3 3 Creative Commons Attribution 3.0 Unported Licence. DEA, CAFT, CDAT, CDET, CDFT and CFIT, have been identified with LC/MS analysis. This study offers DOI: 10.1039/c6ra26918d a cost-effective way to probe the degradation mechanism of atrazine in an aqueous medium by Fenton www.rsc.org/advances oxidation technology. Introduction continues to last for several years. The search for effective methods to remove ATZ from water is of importance. Triazine is used worldwide to combat grassy and broadleaf Water treatment processes include physical processes,12,13 – weeds in many agricultural crops as well as for non-agricultural biological processes,14 16 chemical processes,17,18 catalytic 19,20 This article is licensed under a purposes, such as soil sterilization and road maintenance.1 oxidation and several advanced oxidation processes (AOPs). -

Download Article (PDF)
Advances in Social Science, Education and Humanities Research, volume 336 5th International Conference on Social Science and Higher Education (ICSSHE 19) Research on the Intelligent Old-age Care Mode Based on the Combination of Medical and Health Care Under the Perspective of "Internet +" Yongsheng Hu Min Ren* School of Information Engineering School of Information Engineering Binzhou University Binzhou University Binzhou, Shandong, China Binzhou, Shandong, China [email protected] [email protected] Abstract—This paper summarizes the development status of old-age care in Shandong province, compares and analyzes the II. SUMMARY OF RESEARCH STATUS AT HOME AND advantages of three mainstream old-age care modes, and ABROAD demonstrates the feasibility of "Internet + " smart old-age care mode by taking the combination of "Internet +" and medical and A. Status of Domestic Research nursing care as the starting point, and Puts forward to construct Modern pension service modes mainly include: families, the intelligent endowment innovation mode which ADAPTS to communities, institutions, migratory birds, tourism, and Shandong regional economic development. Guided by the housing pensions[2]. Lu Hongping believes that pension modes regional economy of Shandong Province, the company has can be divided into two types, namely family pension and established the concept of “combination of medical and health [3] care” wisdom and pension, and proposed development strategies social pension . The former is living at home, relying on their such as policy regulation, restructuring and optimization of children to support. The latter is a form of centralized pension medical and pension resources, talent team building, information in fixed places and a commercial activity of welfare nature. -

Understanding the Catalytic Role of Oxalic Acid in the SO3 Hydration To
Understanding the catalytic role of oxalic acid in the SO3 hydration to form H2SO4 in the atmosphere Guochun Lv1, Xiaomin Sun1,*, Chenxi Zhang2, Mei Li3,4* 1Environment Research Institute, Shandong University, Jinan 250100, China 5 2College of Biological and Environmental Engineering, Binzhou University, Binzhou 256600, China 3Institute of Mass Spectrometer and Atmospheric Environment, Jinan University, Guangzhou 510632, China 4Guangdong Provincial Engineering Research Center for on-line source apportionment system of air pollution, Guangzhou 510632, China 10 *Corresponding authors: Xiaomin Sun ([email protected]); Mei Li ([email protected]) Abstract: The hydration of SO3 plays an important role in atmospheric sulfuric acid formation. Some atmospheric species can be involved in and facilitate the reaction. In this work, using quantum chemical calculations, we show that oxalic acid, the most common dicarboxylic acid in the atmosphere, can effectively catalyze the hydration of SO3. The energy barrier of -1 15 SO3 hydration reaction catalyzed by oxalic acid (cTt, tTt, tCt and cCt conformers) is a little higher or less than 1 kcal mol , -1 which is lower than the energy barrier of 5.17 kcal mol for water-catalyzed SO3 hydration. Compared with the rates of SO3 hydration reaction catalyzed by oxalic acid and water, it can be found that, in the upper troposphere, the oxalic acid-catalyzed SO3 hydration can play an important role in promoting the SO3 hydration. It leads us to conclude that the involvement of oxalic acid in SO3 hydration to form H2SO4 is significant in the atmosphere. 20 1. Introduction In the atmosphere, hydrogen atom transfer (HAT) reactions play a significant role in many processes. -
1 Please Read These Instructions Carefully
PLEASE READ THESE INSTRUCTIONS CAREFULLY. MISTAKES IN YOUR CSC APPLICATION COULD LEAD TO YOUR APPLICATION BEING REJECTED. Visit http://studyinchina.csc.edu.cn/#/login to CREATE AN ACCOUNT. • The online application works best with Firefox or Internet Explorer (11.0). Menu selection functions may not work with other browsers. • The online application is only available in Chinese and English. 1 • Please read this page carefully before clicking on the “Application online” tab to start your application. 2 • The Program Category is Type B. • The Agency No. matches the university you will be attending. See Appendix A for a list of the Chinese university agency numbers. • Use the + by each section to expand on that section of the form. 3 • Fill out your personal information accurately. o Make sure to have a valid passport at the time of your application. o Use the name and date of birth that are on your passport. Use the name on your passport for all correspondences with the CLIC office or Chinese institutions. o List Canadian as your Nationality, even if you have dual citizenship. Only Canadian citizens are eligible for CLIC support. o Enter the mailing address for where you want your admission documents to be sent under Permanent Address. Leave Current Address blank. Contact your home or host university coordinator to find out when you will receive your admission documents. Contact information for you home university CLIC liaison can be found here: http://clicstudyinchina.com/contact-us/ 4 • Fill out your Education and Employment History accurately. o For Highest Education enter your current degree studies. -
CONICYT Ranking Por Disciplina > Sub-Área OECD (Académicas) Comisión Nacional De Investigación 1
CONICYT Ranking por Disciplina > Sub-área OECD (Académicas) Comisión Nacional de Investigación 1. Ciencias Naturales > 1.3 Ciencias Físicas y Astronomía Científica y Tecnológica PAÍS INSTITUCIÓN RANKING PUNTAJE FRANCE Universite Paris Saclay (ComUE) 1 5,000 USA University of California Berkeley 2 5,000 USA California Institute of Technology 3 5,000 USA Massachusetts Institute of Technology (MIT) 4 5,000 USA Harvard University 5 5,000 USA Stanford University 6 5,000 UNITED KINGDOM University of Cambridge 7 5,000 FRANCE Sorbonne Universite 8 5,000 USA University of Chicago 9 5,000 JAPAN University of Tokyo 10 5,000 UNITED KINGDOM University of Oxford 11 5,000 FRANCE Universite Sorbonne Paris Cite-USPC (ComUE) 12 5,000 FRANCE University of Paris Diderot 13 5,000 FRANCE PSL Research University Paris (ComUE) 14 5,000 USA Princeton University 15 5,000 FRANCE Universite Paris Sud - Paris XI 16 5,000 CHINA Tsinghua University 17 5,000 USA University of Maryland College Park 18 5,000 UNITED KINGDOM University College London 19 5,000 UNITED KINGDOM Imperial College London 20 5,000 FRANCE Communaute Universite Grenoble Alpes 21 5,000 USA University of Michigan 22 5,000 CANADA University of Toronto 23 5,000 FRANCE Universite Grenoble Alpes (UGA) 24 5,000 ITALY Sapienza University Rome 25 5,000 ITALY University of Padua 26 5,000 CHINA Peking University 27 5,000 UNITED KINGDOM University of Edinburgh 28 5,000 USA University of Illinois Urbana-Champaign 29 5,000 USA Columbia University 30 5,000 INDIA Indian Institute of Technology System (IIT System)