Proteinuria and Progressive Kidney Failure Due to an Inborn Error of Metabolism: Answers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cystinosis, Has Been Reported Previomly in 3 Patients Using (1
, PKU GENE - WSSIBLE CAUSE OF NON-SPECIFIC UENTAL RE- RARE PHENOTYPES OF PLACENTAL ALKALINE PHOSPHATASE: AN 523 TARDATION. Atsuko Fujimoto and Samuel P. Bes-n, ANALYSIS OF RELATIONSHIPS WITH SOME NEONATAL AND b USC Hed. Sch., Dept. Pediatrics, Los Angeles MATERNAL VARIABLES. F. Gloria-Bottini, A. Polzonetti, The Justification Hypothesis (J. Ped. 81:834, 1972) proposes . Bentivoglia, P. Lucarelli and E. Bottini (Spon. by C.D. Cook). that deficiencies in non-essential amino acids might cause mental Jniv. of Camerino, Dept. of Genetics and Computer Center and retardation. The mother heterozygous for synthesis of any one of Jniv. of Rome, Dept. of Pediatrics. the non-essential amino acids would deprive her fetus partially The large number (>15) and frequency (-2%) of rare placental and the heterozygous or homozygous fetus would be more or less alkaline phosphatase (PI) alleles represent a very special case unable to make up for the deficiency. Berman and Ford (Lancet i: among polymorphic enzymes. Since the PI gene is active only dur- 767, 1977) showed that such concatenation of heterozygous mother ing intrauterine life, the allelic diversity and its maintenance and heterozygous fetus is associated with significantly lower IQ. nay be connected with intrauterine environment and with fetal Our own wark has verified this finding. The possibility that development. 1700 newborn infants ( 1271 Caucasians, 337 Negroes heterozygosity for PKU in mother and fetus might be a cause of a and 92 Puerto Ricans), collected at Yale-New Haven Hospital from 1arq.e amount of "non-specific" mental retardation was tested by 1968-1971, were studied. An analysis of the relationship between looking for associated heterozygosity for PKU in mother and child rare PI phenotype and the following 14variableswas carried out: among.12 families in a genetic clinic. -

Effect of Propionic Acid on Fatty Acid Oxidation and U Reagenesis
Pediat. Res. 10: 683- 686 (1976) Fatty degeneration propionic acid hyperammonemia propionic acidemia liver ureagenesls Effect of Propionic Acid on Fatty Acid Oxidation and U reagenesis ALLEN M. GLASGOW(23) AND H. PET ER C HASE UniversilY of Colorado Medical Celller, B. F. SlOlillsky LaboralOries , Denver, Colorado, USA Extract phosphate-buffered salin e, harvested with a brief treatment wi th tryps in- EDTA, washed twice with ph os ph ate-buffered saline, and Propionic acid significantly inhibited "CO z production from then suspended in ph os ph ate-buffe red saline (145 m M N a, 4.15 [I-"ejpalmitate at a concentration of 10 11 M in control fibroblasts m M K, 140 m M c/, 9.36 m M PO" pH 7.4) . I n mos t cases the cells and 100 11M in methyl malonic fibroblasts. This inhibition was we re incubated in 3 ml phosph ate-bu ffered sa lin e cont aining 0.5 similar to that produced by 4-pentenoic acid. Methylmalonic acid I1Ci ll-I4Cj palm it ate (19), final concentration approximately 3 11M also inhibited ' 'C0 2 production from [V 'ejpalmitate, but only at a added in 10 II I hexane. Increasing the amount of hexane to 100 II I concentration of I mM in control cells and 5 mM in methyl malonic did not impair palmit ate ox id ation. In two experiments (Fig. 3) the cells. fibroblasts were in cub ated in 3 ml calcium-free Krebs-Ringer Propionic acid (5 mM) also inhibited ureagenesis in rat liver phosphate buffer (2) co nt ain in g 5 g/ 100 ml essent iall y fatty ac id slices when ammonia was the substrate but not with aspartate and free bovine se rum albumin (20), I mM pa lm itate, and the same citrulline as substrates. -

Incidence of Inborn Errors of Metabolism by Expanded Newborn
Original Article Journal of Inborn Errors of Metabolism & Screening 2016, Volume 4: 1–8 Incidence of Inborn Errors of Metabolism ª The Author(s) 2016 DOI: 10.1177/2326409816669027 by Expanded Newborn Screening iem.sagepub.com in a Mexican Hospital Consuelo Cantu´-Reyna, MD1,2, Luis Manuel Zepeda, MD1,2, Rene´ Montemayor, MD3, Santiago Benavides, MD3, Hector´ Javier Gonza´lez, MD3, Mercedes Va´zquez-Cantu´,BS1,4, and Hector´ Cruz-Camino, BS1,5 Abstract Newborn screening for the detection of inborn errors of metabolism (IEM), endocrinopathies, hemoglobinopathies, and other disorders is a public health initiative aimed at identifying specific diseases in a timely manner. Mexico initiated newborn screening in 1973, but the national incidence of this group of diseases is unknown or uncertain due to the lack of large sample sizes of expanded newborn screening (ENS) programs and lack of related publications. The incidence of a specific group of IEM, endocrinopathies, hemoglobinopathies, and other disorders in newborns was obtained from a Mexican hospital. These newborns were part of a comprehensive ENS program at Ginequito (a private hospital in Mexico), from January 2012 to August 2014. The retrospective study included the examination of 10 000 newborns’ results obtained from the ENS program (comprising the possible detection of more than 50 screened disorders). The findings were the following: 34 newborns were confirmed with an IEM, endocrinopathies, hemoglobinopathies, or other disorders and 68 were identified as carriers. Consequently, the estimated global incidence for those disorders was 3.4 in 1000 newborns; and the carrier prevalence was 6.8 in 1000. Moreover, a 0.04% false-positive rate was unveiled as soon as diagnostic testing revealed negative results. -

Novel Insights Into the Pathophysiology of Kidney Disease in Methylmalonic Aciduria
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2017 Novel Insights into the Pathophysiology of Kidney Disease in Methylmalonic Aciduria Schumann, Anke Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-148531 Dissertation Published Version Originally published at: Schumann, Anke. Novel Insights into the Pathophysiology of Kidney Disease in Methylmalonic Aciduria. 2017, University of Zurich, Faculty of Medicine. Novel Insights into the Pathophysiology of Kidney Disease in Methylmalonic Aciduria Dissertation zur Erlangung der naturwissenschaftlichen Doktorwürde (Dr. sc. nat.) vorgelegt der Mathematisch-naturwissenschaftlichen Fakultät der Universität Zürich von Anke Schumann aus Deutschland Promotionskommission Prof. Dr. Olivier Devuyst (Vorsitz und Leitung der Dissertation) Prof. Dr. Matthias R. Baumgartner Prof. Dr. Stefan Kölker Zürich, 2017 DECLARATION I hereby declare that the presented work and results are the product of my own work. Contributions of others or sources used for explanations are acknowledged and cited as such. This work was carried out in Zurich under the supervision of Prof. Dr. O. Devuyst and Prof. Dr. M.R. Baumgartner from August 2012 to August 2016. Peer-reviewed publications presented in this work: Haarmann A, Mayr M, Kölker S, Baumgartner ER, Schnierda J, Hopfer H, Devuyst O, Baumgartner MR. Renal involvement in a patient with cobalamin A type (cblA) methylmalonic aciduria: a 42-year follow-up. Mol Genet Metab. 2013 Dec;110(4):472-6. doi: 10.1016/j.ymgme.2013.08.021. Epub 2013 Sep 17. Schumann A, Luciani A, Berquez M, Tokonami N, Debaix H, Forny P, Kölker S, Diomedi Camassei F, CB, MK, Faresse N, Hall A, Ziegler U, Baumgartner M and Devuyst O. -

What Disorders Are Screened for by the Newborn Screen?
What disorders are screened for by the newborn screen? Endocrine Disorders The endocrine system is important to regulate the hormones in our bodies. Hormones are special signals sent to various parts of the body. They control many things such as growth and development. The goal of newborn screening is to identify these babies early so that treatment can be started to keep them healthy. To learn more about these specific disorders please click on the name of the disorder below: English: Congenital Adrenal Hyperplasia Esapnol Hiperplasia Suprarrenal Congenital - - http://www.newbornscreening.info/Parents/otherdisorders/CAH.html - http://www.newbornscreening.info/spanish/parent/Other_disorder/CAH.html - Congenital Hypothyroidism (Hipotiroidismo Congénito) - http://www.newbornscreening.info/Parents/otherdisorders/CH.html - http://www.newbornscreening.info/spanish/parent/Other_disorder/CH.html Hematologic Conditions Hemoglobin is a special part of our red blood cells. It is important for carrying oxygen to the parts of the body where it is needed. When people have problems with their hemoglobin they can have intense pain, and they often get sick more than other children. Over time, the lack of oxygen to the body can cause damage to the organs. The goal of newborn screening is to identify babies with these conditions so that they can get early treatment to help keep them healthy. To learn more about these specific disorders click here (XXX). - Sickle Cell Anemia (Anemia de Célula Falciforme) - http://www.newbornscreening.info/Parents/otherdisorders/SCD.html - http://www.newbornscreening.info/spanish/parent/Other_disorder/SCD.html - SC Disease (See Previous Link) - Sickle Beta Thalassemia (See Previous Link) Enzyme Deficiencies Enzymes are special proteins in our body that allow for chemical reactions to take place. -
Blueprint Genetics Hyperammonemia and Urea Cycle Disorder Panel
Hyperammonemia and Urea Cycle Disorder Panel Test code: ME1601 Is a 49 gene panel that includes assessment of non-coding variants. Is ideal for patients with hyperammonemia or a clinical suspicion of a disorder of urea cycle metabolism. The genes on this panel are included in the Comprehensive Metabolism Panel. About Hyperammonemia and Urea Cycle Disorder Congenital urea cycle disorders are the result of defects in the metabolism of nitrogen waste. Deficiency of any of the enzymes in the urea cycle results in an excess of ammonia or other precursor metabolites in the blood. Normally, urea production lowers the ammonia levels in the blood but in the case of defective enzymes, the urea cycle is disturbed. Infants with urea cycle disorders (UCDs) develop cerebral edema, lethargy, hypothermia, neurologic signs and coma, often shortly after birth. Partial, or milder, UCDs are possible if the affected enzyme is positioned in a later phase of the urea cycle. Patients with UCDs may present with hyperammonemia often triggered by stress or illness. The most common primary hyperammonemia is X-linked recessive ornithine transcarbamylase deficiency caused by mutations in the OTC gene. The estimated prevalence is 1:56,000. Prevalence estimates for the other specific urea cycle disorders are 1:200,000 for ASL- and ASS1-related deficiencies and <1:1,000,000 for ARG1, CPS1 and NAGS-related deficiencies. The diagnostic yield ranges from 50% to 80% for different primary urea cycle disorders. In addition to congenital UCDs, this panel has the ability to diagnose other diseases of early phase hyperammonemia and other inborn errors of metabolism showing similar and overlapping symptoms. -

Soonerstart Automatic Qualifying Syndromes and Conditions
SoonerStart Automatic Qualifying Syndromes and Conditions - Appendix O Abetalipoproteinemia Acanthocytosis (see Abetalipoproteinemia) Accutane, Fetal Effects of (see Fetal Retinoid Syndrome) Acidemia, 2-Oxoglutaric Acidemia, Glutaric I Acidemia, Isovaleric Acidemia, Methylmalonic Acidemia, Propionic Aciduria, 3-Methylglutaconic Type II Aciduria, Argininosuccinic Acoustic-Cervico-Oculo Syndrome (see Cervico-Oculo-Acoustic Syndrome) Acrocephalopolysyndactyly Type II Acrocephalosyndactyly Type I Acrodysostosis Acrofacial Dysostosis, Nager Type Adams-Oliver Syndrome (see Limb and Scalp Defects, Adams-Oliver Type) Adrenoleukodystrophy, Neonatal (see Cerebro-Hepato-Renal Syndrome) Aglossia Congenita (see Hypoglossia-Hypodactylia) Aicardi Syndrome AIDS Infection (see Fetal Acquired Immune Deficiency Syndrome) Alaninuria (see Pyruvate Dehydrogenase Deficiency) Albers-Schonberg Disease (see Osteopetrosis, Malignant Recessive) Albinism, Ocular (includes Autosomal Recessive Type) Albinism, Oculocutaneous, Brown Type (Type IV) Albinism, Oculocutaneous, Tyrosinase Negative (Type IA) Albinism, Oculocutaneous, Tyrosinase Positive (Type II) Albinism, Oculocutaneous, Yellow Mutant (Type IB) Albinism-Black Locks-Deafness Albright Hereditary Osteodystrophy (see Parathyroid Hormone Resistance) Alexander Disease Alopecia - Mental Retardation Alpers Disease Alpha 1,4 - Glucosidase Deficiency (see Glycogenosis, Type IIA) Alpha-L-Fucosidase Deficiency (see Fucosidosis) Alport Syndrome (see Nephritis-Deafness, Hereditary Type) Amaurosis (see Blindness) Amaurosis -

Disorders Alphabetical by Disease Updated 1/2020
Disorders Alphabetical by Disease updated 1/2020 Disorders Abbreviation Classification Recommended Uniform Screening Panel (RUSP) Classification 2,4 Dienoyl CoA Reductase Deficiency DE RED Fatty Acid Oxidation Disorder Secondary Condition 2-Methyl 3 Hydroxy Butyric Aciduria 2M3HBA Organic Acid Disorder Secondary Condition 2-Methyl Butyryl-CoA Dehydrogenase Deficiency 2MBG Organic Acid Disorder Secondary Condition (called 2-Methylbutyrylglycinuria on RUSP) 3-Hydroxy-3-Methylglutaryl CoA Lyase Deficiency HMG Organic Acid Disorder Core Condition 3-Methylcrotonyl CoA Carboxylase Deficiency 3MCC Organic Acid Disorder Core Condition 3-Methylglutaconic Aciduria 3MGA Organic Acid Disorder Secondary Condition Alpha-Thalassemia (Bart's Hb) Hemoglobin Bart's Hemoglobin Disorder Secondary Conditoin Argininemia, Arginase Deficiency ARG Amino Acid Disorder Secondary Condition Arginosuccinic Aciduria ASA Amino Acid Disorder Core Condition Benign Hyperphenylalaninemia PHE Amino Acid Disorder Secondary Condition Beta-Ketothiolase Deficiency BKT Organic Acid Disorder Core Condition Biopterin Defect in Cofactor Biosynthesis BIOPT (BS) Amino Acid Disorder Secondary Condition Biopterin Defect in Cofactor Regeneration BIOPT (Reg) Amino Acid Disorder Secondary Condition Biotinidase Deficiency BIO Metabolic Disorder of Biotin Recycling Core Condition Carbamoyltransferase Deficiency, Carbamoyl Phosphate Synthetase I Deficiency CPS Amino Acid Disorder Not on RUSP Carnitine Palmitoyl Transferase Deficiency Type 1 CPT I Fatty Acid Oxidation Disorder Secondary Condition -

List of Disorders Screened
List of Disorders Screened Amino Acid Profile (AA) Disorders Organic Acid Profile (OA) Disorders Argininemia (Arginase Deficiency) *Mitochondrial Acetoacetyl CoA Thiolase (Beta Ketothiolase/SKAT) Carbamoylphosphate Synthethase I Deficiency Deficiency Citrullinemia *Propionic Acidemia *Type I (Arginosuccinate Synthetase Deficiency) *Methylmalonic Acidemia Cobalamin Disorder (CBL A, B) Type II (Citrin Deficiency) Methylmalonic Acidemia Cobalamin Disorder (CBL C, D) *Argininosuccinate Lyase Deficiency (Argininosuccinic Aciduria) *Methylmalonyl-CoA Mutase Deficiency $Nonketotic Hyperglycinemia *Multiple CoA Carboxylase Deficiency due to Glycine Cleavage System H Protein Deficiency Malonic Aciduria (MA) due to Aminomethyltransferase Deficiency Isobutyryl CoA Dehydrogenase Deficiency (IBCD) due to Glycine Decarboxylase Deficiency *Isovaleric Acidemia (IVA) *Homocystinuria or variant forms of Hypermethioninemia 2 Methylbutyryl Glycinuria (2MBG) Hypermethioninemia 2 Methyl 3 Hydroxybutyric Aciduria (2M3HBA) due to Glycine N-Methyltransferase Deficiency *3 Hydroxy 3 Methylglutaryl CoA Lyase Deficiency (HMG) due to S-Adenosylhomocysteine Hydrolase Deficiency *3 Methyl Crotonyl CoA Carboxylase Deficiency (3 MCC) due to Methionine Adenosyltransferase Deficiency 3 Methylglutaconyl CoA Hydratase Deficiency (3MGA) Hyperornithinemia-Hyperammonemia-Homocitrullinuria *Glutaric Acidemia Type I (GAI) Hyperornithinemia-Hyperammonemia-Homocitrullinuria w/gyral atrophy Medium/Short Chain 3 hydroxyacyl CoA dehydrogenase deficiency *Phenylketonuria (M/SCHAD) -

Methylmalonic Acidemia)
NEWBORN SCREENING FACT SHEET MMA (Methylmalonic Acidemia) What is it? respond to vitamin B12 treatment is called mut-. MMA stands for methylmalonic academia. People with mut- type of MMA have too little of It is one type of organic acid disorder. People with the MCM enzyme. MMA have problems breaking down and using certain amino acids and fatty acids from the food Another form of MMA, called MMA with they eat. homocystinuria, is described in a separate fact sheet. See the fact sheet MMA+HCU for more What causes it? information about this condition. In order for the body to use protein from the food we eat, it is broken down into smaller parts called Isoleucine, valine, methionine and threonine are the amino acids. Special enzymes then make changes four amino acids that cannot be used correctly by to the amino acids so the body can use them. In the people with MMA. These amino acids are found in same way, fat from the food we eat is broken down all foods that contain protein. Large amounts are by enzymes into fatty acids that the body can use found in meat, eggs, milk and other dairy products. for energy. Smaller amounts are found in flour, cereal, and some vegetables and fruits. MMA occurs when one of these special enzymes is either missing or not working properly. Without If MMA is not treated, what problems occur? this enzyme, certain amino acids and fatty acids Each child with MMA is likely to have somewhat cannot be used correctly. This causes glycine, different effects. -
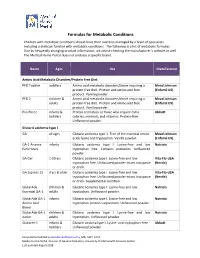
Formulas for Metabolic Conditions
Formulas for Metabolic Conditions Children with metabolic conditions should have their nutrition managed by a team of specialists including a dietician familiar with metabolic conditions. The following is a list of metabolic formulas. Due to frequently changing product information, we advise checking the manufacturer’s website as well. The Medical Home Portal does not endorse a specific brand. Name Ages Use Manufacturer Amino Acid Metabolic Disorders/Protein Free Diet PFD Toddler toddlers Amino acid metabolic disorders/those requiring a Mead Johnson protein‐free diet. Protein and amino acid free (Enfamil US) product. Vanilla powder. PFD 2 children & Amino acid metabolic disorders/those requiring a Mead Johnson adults protein‐free diet. Protein and amino acid free (Enfamil US) product. Vanilla powder. Pro‐Phree infants & Protein restriction or those who require extra Abbott toddlers calories, minerals, and vitamins. Protein‐free. Unflavored powder. Glutaric acidemia type 1 GA all ages Glutaric acidemia type 1. Free of the essential amino Mead Johnson acids lysine and tryptophan. Vanilla powder. (Enfamil US) GA‐1 Anamix infants Glutaric acidemia type I. Lysine‐free and low Nutricia Early Years tryptophan free. Contains prebiotics. Unflavored powder. GA Gel 1‐10 yrs Glutaric acidemia type I. Lysine‐free and low Vita‐Flo‐USA tryptophan free. Unflavored powder mixes into paste (Nestle) or drink. GA Express 15 3 yrs & older Glutaric acidemia type I. Lysine‐free and low Vita‐Flo‐USA tryptophan free. Unflavored powder mixes into paste (Nestle) or drink. Supplemental nutrition. GlutarAde children & Glutaric acidemia type I. Lysine‐free and low Nutricia Essential GA‐1 adults tryptophan. Unflavored powder. -

Methylmalonic Acidemia (Or Aciduria)
orphananesthesia Anaesthesia recommendations for patients suffering from Methylmalonic acidemia (or aciduria) Disease name: Methylmalonic acidemia ICD 10: E71.1 Synonyms: Methylmalonic aciduria, MMA, isolated methylmalonic acidemia Methylmalonic acidemia (MMA) is a group of rare (approx. 1:50,000) autosomal recessive disorders of amino acid metabolism, involving defects in the conversion of methylmalonyl- CoA to succinyl-CoA (which would normally enter the Krebs cycle). The defect is genetically heterogeneous, and can be due to the lack of the enzyme methylmalonyl-CoA mutase 0 - (mut ), partial reduction in its activity (mut ), or to defects in cobalamin metabolism (vit B12 is a cofactor required in the conversion of methylmalonyl-CoA to succinyl-CoA). These defects result in the accumulation of methylmalonic acid. Clinical presentation varies and depends on the genetic diagnosis. The most severe form is mut0, which usually presents in the neonatal period, while other forms may present later in infancy and childhood, when triggers of increased protein catabolism (infection, dehydration, trauma, surgery, stress) cause metabolic decompensation. The presentation may be non- specific, such as vomiting, lethargy and tachypnoea. Neurological manifestations include encephalopathy, seizures, hypotonia and stroke. Gastrointestinal complaints include recurrent vomiting, failure to thrive, and pancreatitis. If the acute crisis is untreated it may progress to coma and death. Medicine in progress Perhaps new knowledge Every patient is unique Perhaps the diagnostic is wrong Find more information on the disease, its centres of reference and patient organisations on Orphanet: www.orpha.net 1 Disease summary Biochemical hallmarks of an acute metabolic decompensation are metabolic acidosis, an increased anion gap (caused by lactate, ketones and organic acids), elevated lactate and ketones ± hyperammonaemia.