A Novel FOXA1/ESR1 Interacting Pathway: a Study of Oncomine™ Breast Cancer Microarrays
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anti-Ps2 / Trefoil Factor 1 Antibody (ARG41084)
Product datasheet [email protected] ARG41084 Package: 100 μl anti-pS2 / Trefoil factor 1 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes pS2 / Trefoil factor 1 Tested Reactivity Hu Tested Application FACS, ICC/IF, IHC-P, IP, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name pS2 / Trefoil factor 1 Antigen Species Human Immunogen Synthetic peptide derived from Human pS2 / Trefoil factor 1. Conjugation Un-conjugated Alternate Names D21S21; pNR-2; Trefoil factor 1; PNR-2; BCEI; hP1.A; HPS2; pS2; Polypeptide P1.A; HP1.A; Protein pS2; Breast cancer estrogen-inducible protein Application Instructions Application table Application Dilution FACS 1:50 ICC/IF 1:50 - 1:200 IHC-P 1:50 - 1:200 IP 1:50 WB 1:500 - 1:2000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 9 kDa Observed Size 11 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.4), 150 mM NaCl, 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol www.arigobio.com 1/2 Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol TFF1 Gene Full Name trefoil factor 1 Background Members of the trefoil family are characterized by having at least one copy of the trefoil motif, a 40-amino acid domain that contains three conserved disulfides. -

Systems and Chemical Biology Approaches to Study Cell Function and Response to Toxins
Dissertation submitted to the Combined Faculties for the Natural Sciences and for Mathematics of the Ruperto-Carola University of Heidelberg, Germany for the degree of Doctor of Natural Sciences Presented by MSc. Yingying Jiang born in Shandong, China Oral-examination: Systems and chemical biology approaches to study cell function and response to toxins Referees: Prof. Dr. Rob Russell Prof. Dr. Stefan Wölfl CONTRIBUTIONS The chapter III of this thesis was submitted for publishing under the title “Drug mechanism predominates over toxicity mechanisms in drug induced gene expression” by Yingying Jiang, Tobias C. Fuchs, Kristina Erdeljan, Bojana Lazerevic, Philip Hewitt, Gordana Apic & Robert B. Russell. For chapter III, text phrases, selected tables, figures are based on this submitted manuscript that has been originally written by myself. i ABSTRACT Toxicity is one of the main causes of failure during drug discovery, and of withdrawal once drugs reached the market. Prediction of potential toxicities in the early stage of drug development has thus become of great interest to reduce such costly failures. Since toxicity results from chemical perturbation of biological systems, we combined biological and chemical strategies to help understand and ultimately predict drug toxicities. First, we proposed a systematic strategy to predict and understand the mechanistic interpretation of drug toxicities based on chemical fragments. Fragments frequently found in chemicals with certain toxicities were defined as structural alerts for use in prediction. Some of the predictions were supported with mechanistic interpretation by integrating fragment- chemical, chemical-protein, protein-protein interactions and gene expression data. Next, we systematically deciphered the mechanisms of drug actions and toxicities by analyzing the associations of drugs’ chemical features, biological features and their gene expression profiles from the TG-GATEs database. -

Harnessing Gene Expression Profiles for the Identification of Ex Vivo Drug
cancers Article Harnessing Gene Expression Profiles for the Identification of Ex Vivo Drug Response Genes in Pediatric Acute Myeloid Leukemia David G.J. Cucchi 1 , Costa Bachas 1 , Marry M. van den Heuvel-Eibrink 2,3, Susan T.C.J.M. Arentsen-Peters 3, Zinia J. Kwidama 1, Gerrit J. Schuurhuis 1, Yehuda G. Assaraf 4, Valérie de Haas 3 , Gertjan J.L. Kaspers 3,5 and Jacqueline Cloos 1,* 1 Hematology, Cancer Center Amsterdam, Amsterdam UMC, Vrije Universiteit Amsterdam, 1081 HV Amsterdam, The Netherlands; [email protected] (D.G.J.C.); [email protected] (C.B.); [email protected] (Z.J.K.); [email protected] (G.J.S.) 2 Department of Pediatric Oncology/Hematology, Erasmus MC–Sophia Children’s Hospital, 3015 CN Rotterdam, The Netherlands; [email protected] 3 Princess Máxima Center for Pediatric Oncology, 3584 CS Utrecht, The Netherlands; [email protected] (S.T.C.J.M.A.-P.); [email protected] (V.d.H.); [email protected] (G.J.L.K.) 4 The Fred Wyszkowski Cancer Research, Laboratory, Department of Biology, Technion-Israel Institute of Technology, 3200003 Haifa, Israel; [email protected] 5 Emma’s Children’s Hospital, Amsterdam UMC, Vrije Universiteit Amsterdam, Pediatric Oncology, 1081 HV Amsterdam, The Netherlands * Correspondence: [email protected] Received: 21 April 2020; Accepted: 12 May 2020; Published: 15 May 2020 Abstract: Novel treatment strategies are of paramount importance to improve clinical outcomes in pediatric AML. Since chemotherapy is likely to remain the cornerstone of curative treatment of AML, insights in the molecular mechanisms that determine its cytotoxic effects could aid further treatment optimization. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Location Analysis of Estrogen Receptor Target Promoters Reveals That
Location analysis of estrogen receptor ␣ target promoters reveals that FOXA1 defines a domain of the estrogen response Jose´ e Laganie` re*†, Genevie` ve Deblois*, Ce´ line Lefebvre*, Alain R. Bataille‡, Franc¸ois Robert‡, and Vincent Gigue` re*†§ *Molecular Oncology Group, Departments of Medicine and Oncology, McGill University Health Centre, Montreal, QC, Canada H3A 1A1; †Department of Biochemistry, McGill University, Montreal, QC, Canada H3G 1Y6; and ‡Laboratory of Chromatin and Genomic Expression, Institut de Recherches Cliniques de Montre´al, Montreal, QC, Canada H2W 1R7 Communicated by Ronald M. Evans, The Salk Institute for Biological Studies, La Jolla, CA, July 1, 2005 (received for review June 3, 2005) Nuclear receptors can activate diverse biological pathways within general absence of large scale functional data linking these putative a target cell in response to their cognate ligands, but how this binding sites with gene expression in specific cell types. compartmentalization is achieved at the level of gene regulation is Recently, chromatin immunoprecipitation (ChIP) has been used poorly understood. We used a genome-wide analysis of promoter in combination with promoter or genomic DNA microarrays to occupancy by the estrogen receptor ␣ (ER␣) in MCF-7 cells to identify loci recognized by transcription factors in a genome-wide investigate the molecular mechanisms underlying the action of manner in mammalian cells (20–24). This technology, termed 17-estradiol (E2) in controlling the growth of breast cancer cells. ChIP-on-chip or location analysis, can therefore be used to deter- We identified 153 promoters bound by ER␣ in the presence of E2. mine the global gene expression program that characterize the Motif-finding algorithms demonstrated that the estrogen re- action of a nuclear receptor in response to its natural ligand. -

S41467-020-16223-7.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-16223-7 OPEN Trefoil factors share a lectin activity that defines their role in mucus Michael A. Järvå 1,2, James P. Lingford1,2, Alan John1,2, Niccolay Madiedo Soler1,2, Nichollas E. Scott 3 & ✉ Ethan D. Goddard-Borger 1,2 The mucosal epithelium secretes a host of protective disulfide-rich peptides, including the trefoil factors (TFFs). The TFFs increase the viscoelasticity of the mucosa and promote cell 1234567890():,; migration, though the molecular mechanisms underlying these functions have remained poorly defined. Here, we demonstrate that all TFFs are divalent lectins that recognise the GlcNAc-α-1,4-Gal disaccharide, which terminates some mucin-like O-glycans. Degradation of this disaccharide by a glycoside hydrolase abrogates TFF binding to mucins. Structural, mutagenic and biophysical data provide insights into how the TFFs recognise this dis- accharide and rationalise their ability to modulate the physical properties of mucus across different pH ranges. These data reveal that TFF activity is dependent on the glycosylation state of mucosal glycoproteins and alludes to a lectin function for trefoil domains in other human proteins. 1 The Walter and Eliza Hall Institute of Medical Research, Parkville, VIC 3052, Australia. 2 Department of Medical Biology, University of Melbourne, Parkville, VIC 3010, Australia. 3 Department of Microbiology and Immunology, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, ✉ Parkville, VIC 3010, Australia. email: [email protected] NATURE COMMUNICATIONS | (2020) 11:2265 | https://doi.org/10.1038/s41467-020-16223-7 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-16223-7 he three human TFFs (TFF1, TFF2, and TFF3)1 are ubi- considering the glycosylation state of mucosal proteins when Tquitous in mucosal environments. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Identification and Characterization of Tissue-Resident Memory T Cells in Humans Brahma Vencel Kumar
Identification and characterization of tissue-resident memory T cells in humans Brahma Vencel Kumar Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2018 ©2017 Brahma Vencel Kumar All rights reserved ABSTRACT Identification and characterization of tissue-resident memory T cells in humans Brahma Vencel Kumar Memory T cells are critical for maintaining lifelong immunity by protecting against reinfection with previously encountered pathogens. In recent years, a subset of memory T cells termed tissue-resident memory T cells (TRM) has emerged as the primary mediator of protection at many tissue sites. Numerous studies in mice have demonstrated that TRM accelerate pathogen clearance compared with other subsets of memory T cells. The defining characteristic of TRM is that they are retained within tissues and do not circulate in the blood. The lack of TRM in blood has proved to be a barrier for investigating the role of TRM in healthy humans. As a result, there are many outstanding questions about TRM biology in humans, including which phenotypic markers identify TRM, if TRM represent a unique memory subset, as well as defining transcriptional and functional characteristics of this subset. Through a unique collaboration with the local organ procurement agency, we obtained samples from >15 tissue sites from healthy organ donors of all ages. We found that the surface marker CD69 was expressed by memory CD4 + and CD8 + T cells in multiple tissues including spleen and other lymphoid tissues, lung, and intestines, but not in blood, suggesting that this marker may identify TRM in human tissues. -

BMC Genomics Biomed Central
BMC Genomics BioMed Central Research article Open Access Differential gene expression in femoral bone from red junglefowl and domestic chicken, differing for bone phenotypic traits Carl-Johan Rubin1, Johan Lindberg2, Carolyn Fitzsimmons3, Peter Savolainen2, Per Jensen4, Joakim Lundeberg2, Leif Andersson3,5 and Andreas Kindmark*1 Address: 1Department of Medical Sciences, Uppsala University, Sweden, 2Department of Gene Technology, School of Biotechnology, Royal Institute of Technology, Stockholm, Sweden, 3Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences, Uppsala, Sweden, 4IFM Biology, Linköping University, SE-585 83, Linköping, Sweden and 5Department of Medical Biochemistry and Microbiology, Uppsala University, Box 597, SE-75124 Uppsala, Sweden Email: Carl-Johan Rubin - [email protected]; Johan Lindberg - [email protected]; Carolyn Fitzsimmons - [email protected]; Peter Savolainen - [email protected]; Per Jensen - [email protected]; Joakim Lundeberg - [email protected]; Leif Andersson - [email protected]; Andreas Kindmark* - [email protected] * Corresponding author Published: 2 July 2007 Received: 2 April 2007 Accepted: 2 July 2007 BMC Genomics 2007, 8:208 doi:10.1186/1471-2164-8-208 This article is available from: http://www.biomedcentral.com/1471-2164/8/208 © 2007 Rubin et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Osteoporosis is frequently observed among aging hens from egg-producing strains (layers) of domestic chicken. -
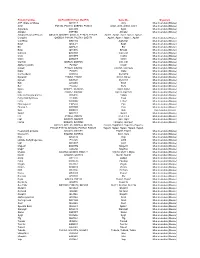
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Origins and Functional Impact of Copy Number Variation in the Human Genome
doi:10.1038/nature08516 ARTICLES Origins and functional impact of copy number variation in the human genome Donald F. Conrad1*, Dalila Pinto2*, Richard Redon1,3, Lars Feuk2,4, Omer Gokcumen5, Yujun Zhang1, Jan Aerts1, T. Daniel Andrews1, Chris Barnes1, Peter Campbell1, Tomas Fitzgerald1, Min Hu1, Chun Hwa Ihm5, Kati Kristiansson1, Daniel G. MacArthur1, Jeffrey R. MacDonald2, Ifejinelo Onyiah1, Andy Wing Chun Pang2, Sam Robson1, Kathy Stirrups1, Armand Valsesia1, Klaudia Walter1, John Wei2, Wellcome Trust Case Control Consortium{, Chris Tyler-Smith1, Nigel P. Carter1, Charles Lee5, Stephen W. Scherer2,6 & Matthew E. Hurles1 Structural variations of DNA greater than 1 kilobase in size account for most bases that vary among human genomes, but are still relatively under-ascertained. Here we use tiling oligonucleotide microarrays, comprising 42 million probes, to generate a comprehensive map of 11,700 copy number variations (CNVs) greater than 443 base pairs, of which most (8,599) have been validated independently. For 4,978 of these CNVs, we generated reference genotypes from 450 individuals of European, African or East Asian ancestry. The predominant mutational mechanisms differ among CNV size classes. Retrotransposition has duplicated and inserted some coding and non-coding DNA segments randomly around the genome. Furthermore, by correlation with known trait-associated single nucleotide polymorphisms (SNPs), we identified 30 loci with CNVs that are candidates for influencing disease susceptibility. Despite this, having assessed the completeness of our map and the patterns of linkage disequilibrium between CNVs and SNPs, we conclude that, for complex traits, the heritability void left by genome-wide association studies will not be accounted for by common CNVs. -

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes.