A Dissertation Entitled Orthometallated Acetophenone Imines As Ligands
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

FAROOK COLLEGE (Autonomous)
FAROOK COLLEGE (Autonomous) M.Sc. DEGREE PROGRAMME IN CHEMISTRY CHOICE BASED CREDIT AND SEMESTER SYSTEM-PG (FCCBCSSPG-2019) SCHEME AND SYLLABI 2019 ADMISSION ONWARDS 1 CERTIFICATE I hereby certify that the documents attached are the bona fide copies of the syllabus of M.Sc. Chemistry Programme to be effective from the academic year 2019-20 onwards. Date: Place: P R I N C I P A L 2 FAROOK COLLEGE (AUTONOMOUS) MSc. CHEMISTRY (CSS PATTERN) Regulations and Syllabus with effect from 2019 admission Pattern of the Programme a. The name of the programme shall be M.Sc. Chemistry under CSS pattern. b. The programme shall be offered in four semesters within a period of two academic years. c. Eligibility for admission will be as per the rules laid down by the College from time to time. d. Details of the courses offered for the programme are given in Table 1. The programme shall be conducted in accordance with the programme pattern, scheme of examination and syllabus prescribed. Of the 25 hours per week, 13 hours shall be allotted for theory and 12 hours for practical; 1 theory hour per week during even semesters shall be allotted for seminar. Theory Courses In the first three semesters, there will be four theory courses; and in the fourth semester, three theory courses. All the theory courses in the first and second semesters are core courses. In the third semester there will be three core theory courses and one elective theory course. College can choose any one of the elective courses given in Table 1. -

Download Article
Volume 10, Issue 3, 2020, 5412 - 5417 ISSN 2069-5837 Biointerface Research in Applied Chemistry www.BiointerfaceResearch.com https://doi.org/10.33263/BRIAC103.412417 Original Research Article Open Access Journal Received: 26.01.2020 / Revised: 02.03.2020 / Accepted: 03.03.2020 / Published on-line: 09.03.2020 Obtaining salts of resin acids from Cuban pine by metathesis reactions Olinka Tiomno-Tiomnova 1 , Jorge Enrique Rodriguez-Chanfrau 2 , Daylin Gamiotea-Turro 2 , Thayná Souza Silva 3 , Carlos Henrique Gomes Martins 3 , Alexander Valerino-Diaz 2 , Yaymarilis Veranes-Pantoja 4 , Angel Seijo-Santos 1 , Antonio Carlos Guastaldi 2 1Center of Engineering and Chemical Investigations. Havana. CP 10600, Cuba 2Group of Biomaterials, Department of Physical Chemistry, Institute of Chemistry, Sao ~ Paulo State University (UNESP), Araraquara, SP, 14800-060, Brazil 3Laboratory of Research on Antimicrobial Trials (LaPEA), Institute of Biomedical Sciences – ICBIM, Federal University of Uberlândia, Uberlândia, Brazil 4Biomaterials Center. University of Havana. Havana. CP 10600, Cuba *corresponding author e-mail address: [email protected] | Scopus ID 7801488065 ABSTRACT The resin acids obtained from the Cuban pine rosin are the starting material for the development and application of metathesis reactions. These reactions allow the obtaining of salts with high added value which could be used in the development of biomaterials for dental use. The objective of this work was to obtain calcium, magnesium and zinc resinates from the resin acid obtained from the Cuban pine resin by metathesis reaction for possible use in the development of biomaterials. The products obtained were evaluated by elemental analysis, X-ray powder diffraction, infrared spectroscopy, scanning electron microscopy associated with electron dispersion spectroscopy, nuclear magnetic resonance and biological tests (antibacterial activity). -

A Metal-Free Approach to Biaryl Compounds: Carbon-Carbon Bond Formation from Diaryliodonium Salts and Aryl Triolborates
Portland State University PDXScholar Dissertations and Theses Dissertations and Theses Winter 4-3-2015 A Metal-Free Approach to Biaryl Compounds: Carbon-Carbon Bond Formation from Diaryliodonium Salts and Aryl Triolborates Kuruppu Lilanthi Jayatissa Portland State University Follow this and additional works at: https://pdxscholar.library.pdx.edu/open_access_etds Part of the Catalysis and Reaction Engineering Commons Let us know how access to this document benefits ou.y Recommended Citation Jayatissa, Kuruppu Lilanthi, "A Metal-Free Approach to Biaryl Compounds: Carbon-Carbon Bond Formation from Diaryliodonium Salts and Aryl Triolborates" (2015). Dissertations and Theses. Paper 2229. https://doi.org/10.15760/etd.2226 This Thesis is brought to you for free and open access. It has been accepted for inclusion in Dissertations and Theses by an authorized administrator of PDXScholar. Please contact us if we can make this document more accessible: [email protected]. A Metal-Free Approach to Biaryl Compounds: Carbon-Carbon Bond Formation from Diaryliodonium Salts and Aryl Triolborates. by Kuruppu Lilanthi Jayatissa A thesis submitted in partial fulfillment of the requirement for the degree of Master of Science in Chemistry Thesis Committee: David Stuart, Chair Robert Strongin Theresa McCormick Portland State University 2015 Abstract Biaryl moieties are important structural motifs in many industries, including pharmaceutical, agrochemical, energy and technology. The development of novel and efficient methods to synthesize these carbon-carbon bonds is at the forefront of synthetic methodology. Since Ullmann’s first report of stoichiometric Cu-mediated homo-coupling of aryl halides, there has been a dramatic evolution in transition metal catalyzed biaryl cross-coupling reactions. -

Organometallic Compounds
Chapter 11 Organometallic compounds Organometallics Reactions using organometallics Organometallic compounds Ch 11 #2 = comp’s containing a carbon-metal bond δ− δ+ C in organometallic comp’ds are nucleophilic. C in organic comp’d (like ROH, RNH , and RX) 2 δ+ δ are electrophilic. − due to ∆EN Ch 11 #3 in substitution reactions a carbon Nu: R-Li and R-MgX Ch 11 #4 (used to be) the two most common organometallics organolithium comp’ds BuLi = an alkyl lithium organomagnesium comp’ds = Grignard reagents 1912 Nobel Prize R, Ar, vinyl all possible; Br (as X) popular Ether (solvent) coordinates Mg, stabilizing it. Reactions of R-Li and R-MgX Ch 11 #5 reacts like a carbanion C:– ~ a C Nu: reactions as C Nu: (like SN(2)) nucleophilic addition to carbonyls ~ more often Chapt 16 Ch 11 #6 R-Li and R-MgX are very strong B: how strong? pKa? − − react even with very weak acid stronger than OH? NH2? useful for preparing deuterated HC Storage and reaction must be acid- and moisture-free. Transmetal(l)ation Ch 11 #7 R-Li is more reactive than R-MgX is. C–Li more polar than C–Mg C of R-Li more nu-philic [better Nu:] transmetalation [metal exchange] to less reactive [more stable] organometallic Coupling using Gilman reagent Ch 11 #8 coupling reaction (in organic chemistry) two hydrocarbon fragments are coupled (to form C–C) with the aid of a (transition) metal catalyst Gilman reagent coupling of R of R-X and R’ of Gilman reagent RX + R’2CuLi R–R’ mechanism? substitution of X with R’? not clear Ch 11 #9 R can be alkyl, aryl, or alkenyl [vinyl] which is not possible by R-Li or R-MgX Is R of Gilman why? they are SN(2). -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -
Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry
REVIEWS Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry Eiichi Nakamura* and Seiji Mori Organocopper reagents provide the principles. This review will summarize example of molecular recognition and most general synthetic tools in organic first the general structural features of supramolecular chemistry, which chemistry for nucleophilic delivery of organocopper compounds and the pre- chemists have long exploited without hard carbanions to electrophilic car- vious mechanistic arguments, and then knowing it. Reasoning about the bon centers. A number of structural describe the most recent mechanistic uniqueness of the copper atom among and mechanistic studies have been pictures obtained through high-level neighboring metal elements in the reported and have led to a wide variety quantum mechanical calculations for periodic table will be presented. of mechanistic proposals, some of three typical organocuprate reactions, which might even be contradictory to carbocupration, conjugate addition, Keywords: catalysis ´ conjugate addi- others. With the recent advent of and SN2 alkylation. The unified view tions ´ copper ´ density functional physical and theoretical methodolo- on the nucleophilic reactivities of met- calculations ´ supramolecular chemis- gies, the accumulated knowledge on al organocuprate clusters thus ob- try organocopper chemistry is being put tained has indicated that organocup- together into a few major mechanistic rate chemistry represents an intricate 1. Introduction 1 R Cu X The desire to learn about the nature of elements has been R or R1 R and will remain a main concern of chemists. In this review, we R Cu will consider what properties of copper make organocopper R1 chemistry so useful in organic chemistry. -

Acetophenone Sigma Aldrich.Pdf
SIGMA-ALDRICH sigma-aldrich.com Material Safety Data Sheet Version 4.0 Revision Date 02/27/2010 Print Date 07/08/2011 1. PRODUCT AND COMPANY IDENTIFICATION Product name : Acetophenone Product Number : A10701 Brand : Sigma-Aldrich Company : Sigma-Aldrich 3050 Spruce Street SAINT LOUIS MO 63103 USA Telephone : +1 800-325-5832 Fax : +1 800-325-5052 Emergency Phone # : (314) 776-6555 2. HAZARDS IDENTIFICATION Emergency Overview OSHA Hazards Combustible Liquid, Harmful by ingestion., Irritant GHS Label elements, including precautionary statements Pictogram Signal word Danger Hazard statement(s) H227 Combustible liquid H302 Harmful if swallowed. H318 Causes serious eye damage. Precautionary statement(s) P210 Keep away from heat/sparks/open flames/hot surfaces. - No smoking. P264 Wash skin thoroughly after handling. P270 Do not eat, drink or smoke when using this product. P280 Wear protective gloves/protective clothing/eye protection/face protection. P301 + P312 IF SWALLOWED: Call a POISON CENTER or doctor/physician if you feel unwell. P305 + P351 + P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. P310 Immediately call a POISON CENTER or doctor/physician. P330 Rinse mouth. P370 + P378 In case of fire: Use dry sand, dry chemical or alcohol-resistant foam for extinction. P403 + P235 Store in a well-ventilated place. Keep cool. P501 Dispose of contents/container to an approved waste disposal plant. HMIS Classification Health hazard: 2 Flammability: 2 Physical hazards: 0 NFPA Rating Health hazard: 2 Fire: 2 Reactivity Hazard: 0 Potential Health Effects Sigma-Aldrich - A10701 Page 1 of 6 Inhalation May be harmful if inhaled. -

THAT ARE NOT ALLOLLIKULTTUUS009958363B2 (12 ) United States Patent ( 10 ) Patent No
THAT ARE NOT ALLOLLIKULTTUUS009958363B2 (12 ) United States Patent ( 10 ) Patent No. : US 9 , 958 ,363 B2 Smart et al. (45 ) Date of Patent: May 1 , 2018 ( 54 ) FLUOROUS AFFINITY EXTRACTION FOR (56 ) References Cited IONIC LIQUID - BASED SAMPLE PREPARATION U . S . PATENT DOCUMENTS 7 ,273 ,587 B1 9 / 2007 Birkner et al . ( 71 ) Applicant: Agilent Technologies , Inc. , Loveland , 7 ,273 ,720 B1 9 / 2007 Birkner et al. CA (US ) ( Continued ) ( 72 ) Inventors: Brian Phillip Smart, San Jose , CA (US ) ; Brooks Bond - Watts , Fremont, FOREIGN PATENT DOCUMENTS CA (US ) ; James Alexander Apffel, Jr ., WO WO2007110637 10 /2007 Mountain View , CA (US ) WO WO2011155829 12 / 2011 ( 73 ) Assignee : Agilent Technologies , Inc ., Santa Clara , OTHER PUBLICATIONS CA (US ) Yuesheng Ye , Yossef A . Elabd “ Anion exchanged polymerized ionic ( * ) Notice : Subject to any disclaimer, the term of this liquids: High free volume single ion conductors ” Polymer 52 ( 2011 ) patent is extended or adjusted under 35 1309 - 1317 , plus supplementary data ( pp . 1 - 6 ) . * U . S . C . 154 (b ) by 168 days . (Continued ) ( 21) Appl . No. : 14 /724 ,656 Primary Examiner — Christine T Mui ão( 22 ) Filed : May 28 , 2015 Assistant Examiner - Emily R . Berkeley (6565 ) Prior Publication Data (57 ) ABSTRACT US 2015 /0369711 A1 Dec . 24 , 2015 A method for removing an ionic liquid from an aqueous Related U . S . Application Data sample is provided . In some embodiments , the method includes : ( a ) combining an aqueous sample including an ( 60 ) Provisional application No . 62/ 016 ,000 , filed on Jun . ionic liquid with an ion exchanger composition including an 23 , 2014 , provisional application No . 62 /016 , 003 , ion exchanger counterion to produce a solution including a (Continued ) fluorous salt of the ionic liquid , where at least one of the ionic liquid and the ion exchanger counterion is fluorinated ; (51 ) Int . -

1 Structural Characterization of a Tetrametallic Diamine-Bis(Phenolate) Complex of Lithium and Synthesis of a Related Bismuth Co
Structural Characterization of a Tetrametallic Diamine-bis(phenolate) Complex of Lithium and Synthesis of a Related Bismuth Complex Marcus W. Drover,a Jennifer N. Murphy,a Jenna C. Flogeras,a Céline M. Schneider,a,b Louise N. Dawe,a,c and Francesca M. Kerton*a a Department of Chemistry, Memorial University of Newfoundland St. John’s, Newfoundland, Canada A1B 3X7 b NMR Laboratory, C-CART, Memorial University of Newfoundland, St. John’s, NL, A1B 3X7, Canada c C-CART X-ray Diffraction Laboratory, Memorial University of Newfoundland, St. John’s, Newfoundland, Canada A1B 3X7; Current address, Department of Chemistry, Wilfrid Laurier University, Science Building, 75 University Ave. W., Waterloo, Ontario, Canada N2L 3C5 * Author to whom correspondence should be addressed. Tel: +1-709-864-8089, Fax: +1-709- 864-3702, E-mail: [email protected] Contribution For Special Issue on “Modern Canadian Inorganic Chemistry” Abstract A novel lithium complex was prepared from the reaction of 1,4-bis(2-hydroxy-3,5-di-tert-butyl- BuBuIm benzyl)imidazolidine H2[O2N2] (L1H2) with n-butyllithium to provide the corresponding 7 tetralithium amine-bis(phenolate) complex {Li2[L1]}2•4THF, 1. Variable temperature Li NMR revealed that this complex is labile in solution, dissociating at elevated temperatures to afford two dilithium entities. Additionally, 7Li MAS NMR was performed on 1 to provide information regarding the lithium coordination environment in the bulk solid-state. The reactivity of 1 was assessed in the ring-expansion polymerization of ε-caprolactone (ε-CL), which was first order in ε-CL with an activation energy of 50.9 kJmol-1. -

ACETOPHENONE Method No. PV2003 Target Concentration
ACETOPHENONE Method no. PV2003 Target concentration: 100 ppm (491 mg/m3) Procedure: Samples are collected by drawing a known volume of air through a Tenax GC tube. Samples are desorbed with a solvent mixture of (5:95) Isopropanol:Carbon Disulfide (IPA):(CS2) and analyzed by gas chromatography with a flame ionization detector. Recommended air volume 120 minutes at 0.1 L/min (12 L) and sampling rate: Status of Method: Partially Validated. This method has been only partially evaluated and is presented for information and trial use. July 1982 Wayne Potter Service Branch 1 OSHA Salt Lake Technical Center Salt Lake City UT-84115 1 of 8 1 General Discussion 1.1 Background 1.1.1 History of procedure Recently, the OSHA Analytical Laboratory received a set of field samples that required analysis for acetophenone. The air samples had been collected on charcoal tubes and isopropanol impingers. Desorption studies were done on charcoal tubes using acetone, carbon disulfide, 5:95 isopropanol:carbon disulfide, and methylene chloride as desorbing solvents. The best results were obtained with 5:95 isopropanol:carbon disulfide, but the recovery was only 68%, which indicated that charcoal tubes should not be recommended for the collection of acetophenone air samples. An evaluation of isopropanol impingers was not performed because it was preferable to find an adsorbent procedure for sample collection. NIOSH method 291 for α -chloroacetophenone uses Tenax GC tubes. The Tenax GC tubes were tried for acetophenone, and appeared to be a suitable method of collection. 1.1.2 Toxic effects (This section is for information only and should not be taken as the basis of OSHA policy.) Acetophenone is a narcotic in high concentrations (Ref. -
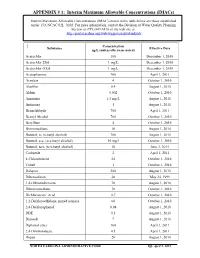
APPENDIX # 1: Interim Maximum Allowable Concentrations (Imacs)
APPENDIX # 1: Interim Maximum Allowable Concentrations (IMACs) Interim Maximum Allowable Concentrations (IMAC) shown in the table below are those established under 15A NCAC 02L .0202. For more information, contact the Division of Water Quality Planning Section at (919) 807-6416 or the web site at: http://portal.ncdenr.org/web/wq/ps/csu/gwstandards Concentration Substance Effective Date ug/L (unless otherwise noted) Acetochlor 100 December 1, 2010 Acetochlor ESA 1 mg/L December 1, 2010 Acetochlor OXA 1 mg/L December 1, 2010 Acetophenone 700 April 1, 2011 Acrolein 4 October 1, 2010 Alachlor 0.4 August 1, 2010 Aldrin 0.002 October 1, 2010 Ammonia 1.5 mg/L August 1, 2010 Antimony 1 August 1, 2010 Benzaldehyde 700 April 1, 2011 Benzyl Alcohol 700 October 1, 2010 Beryllium 4 October 1, 2010 Bromomethane 10 August 1, 2010 Butanol, n- (n-butyl alcohol) 700 August 1, 2010 Butanol, sec- (sec-butyl alcohol) 10 mg/l October 1, 2010 Butanol, tert- (tert-butyl alcohol) 10 June 1, 2011 Carbazole 2 April 1, 2011 4-Chlorotoluene 24 October 1, 2010 Cobalt 1 October 1, 2010 Dalapon 200 August 1, 2010 Dibenzofuran 28 May 24, 1999 1,4-Dibromobenzene 70 August 1, 2010 Dibromomethane 70 October 1, 2010 Dichloroacetic Acid 0.7 October 1, 2010 1,2-Dichloroethylene, mixed isomers 60 October 1, 2010 2,4-Dichlorophenol 0.98 August 1, 2010 DDE 0.1 August 1, 2010 Dinoseb 7 August 1, 2010 Diphenyl ether 100 April 1, 2011 2,4-Dinitrotoluene 0.1 April 1, 2011 Diquat 20 August 1, 2010 NORTH CAROLINA ADMINISTRATIVE CODE Eff. -

Unhindered Triangulene Salt Pairs: Substitution- Dependent Contact Ion Pairing and Complex Solvent- Separated Discotic Ions in Solution
University of Kentucky UKnowledge Theses and Dissertations--Chemistry Chemistry 2015 UNHINDERED TRIANGULENE SALT PAIRS: SUBSTITUTION- DEPENDENT CONTACT ION PAIRING AND COMPLEX SOLVENT- SEPARATED DISCOTIC IONS IN SOLUTION Subrahmanyam Modekrutti University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Modekrutti, Subrahmanyam, "UNHINDERED TRIANGULENE SALT PAIRS: SUBSTITUTION-DEPENDENT CONTACT ION PAIRING AND COMPLEX SOLVENT-SEPARATED DISCOTIC IONS IN SOLUTION" (2015). Theses and Dissertations--Chemistry. 50. https://uknowledge.uky.edu/chemistry_etds/50 This Doctoral Dissertation is brought to you for free and open access by the Chemistry at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Chemistry by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. I agree that the document mentioned above may be made available immediately for worldwide access unless an embargo applies.