Explaining Large Mitochondrial Sequence Differences Within A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Development and Investigation of an Audio Lure for Improved Possum (Trichosurus Vulpecula) Monitoring and Control in New Zealand
Lincoln University Digital Thesis Copyright Statement The digital copy of this thesis is protected by the Copyright Act 1994 (New Zealand). This thesis may be consulted by you, provided you comply with the provisions of the Act and the following conditions of use: you will use the copy only for the purposes of research or private study you will recognise the author's right to be identified as the author of the thesis and due acknowledgement will be made to the author where appropriate you will obtain the author's permission before publishing any material from the thesis. The Possum Pied Piper: the development and investigation of an audio lure for improved possum (Trichosurus vulpecula) monitoring and control in New Zealand A thesis submitted in partial fulfilment of the requirements for the Degree of Doctor of Philosophy at Lincoln University by Matthew J. Kavermann Lincoln University 2013 ii iii Declaration Some aspects of this thesis have been published or accepted for publication (copies of the published and submitted papers are attached at the back of the thesis) or presented at conferences. Publications Kavermann M, Ross J, Paterson A, Eason, C. (in press) Progressing the possum pied piper project. Proceedings of the 25th Vertebrate Pest Conference, Monterey Ca 2012. Dilks P, Shapiro L, Greene T, Kavermann M, Eason CT, Murphy EC (2011). Field evaluation of para- aminopropiophenone (PAPP) for controlling stoats (Mustela erminea) in New Zealand. New Zealand Journal of Zoology 38(2): 143-150 Conference presentations Kavermann M, Ross J, Paterson A, Harper, G. 2012 Assessing the sensitivity of interference based monitoring devices to possum presence. -

Hemideina Crassidens
UNIVERSITÉ DU QUÉBEC À MONTRÉAL LE MAINTIEN DE STRATÉGIES ALTERNATIVES CHEZ LE WELLINGTON TREE WETA (HEM/DE/NA CRASSIDENS): TESTER LES PRÉDICTIONS DE L'HYPOTHÈSE DE POLYMORPHISME GÉNÉTIQUE MÉMOIRE PRÉSENTÉ COMME EXIGENCE PARTIELLE DE LA MAÎTRISE EN BIOLOGIE PAR SARAHNASON JANVIER 2019 UNIVERSITÉ DU QUÉBEC À MONTRÉAL Service des bibliothèques Avertissement La diffusion de ce mémoire se fait dans le respect des droits de son auteur, qui a signé le formulaire Autorisation de reproduire et de diffuser un travail de recherche de cycles supérieurs (SDU-522 - Rév.07-2011). Cette autorisation stipule que «conformément à l'article 11 du Règlement no 8 des études de cycles supérieurs, [l'auteur] concède à l'Université du Québec à Montréal une licence non exclusiye d'utilisation et de publication de la totalité ou d'une partie importante de [son] travail de recherche pour des fins pédagogiques et non commerciales. Plus précisément, [l'auteur] autorise l'Université du Québec à Montréal à reproduire, diffuser, prêter, distribuer ou vendre des copies de [son] travail de recherche à des fins non commerciales sur quelque support que ce soit, y compris l'Internet. Cette licence et cette autorisation n'entraînent pas une renonciation de [la] part [de l'auteur] à [ses] droits moraux ni à [ses] droits de propriété intellectuelle. Sauf entente contraire, [l'auteur] conserve la liberté de diffuser et de commercialiser ou non ce travail dont [il] possède un exemplaire.» UNIVERSITÉ DU QUÉBEC À MONTRÉAL THE MAINTENANCE OF ALTERNATIVE STRATEGIES IN WELLINGTON TREE WETA (HEMIDEINA CRASSJDENS): TESTING THE PREDICTIONS OF THE GENETIC POLYMORPHISM HYPOTHESIS MÉMOIRE PRESENTED IN FULFILLMENT OF MASTER'S IN BIOLOGY BY SARAHNASON JANUARY, 2019 REMERCIEMENTS 1 would like to thank my supervisor Dr. -

Male Tree Weta Are Attracted to Cuticular Scent Cues but Do Not Discriminate According to Sex Or Among Two Closely Related Species
Received: 26 April 2017 | Revised: 7 June 2017 | Accepted: 3 July 2017 DOI: 10.1111/eth.12652 RESEARCH PAPER Male tree weta are attracted to cuticular scent cues but do not discriminate according to sex or among two closely related species Priscilla M. Wehi1,2 | Adrian Monks2 | Mary Morgan-Richards1 1Institute of Agriculture & Environment, Massey University, Palmerston North, Abstract New Zealand Recognition of conspecifics is an essential precursor of successful mating. Where 2 Landcare Research, Dunedin, New Zealand related species coexist, species discrimination might be important, but because related Correspondence species are similar, species signal recognition may actually be low. Chemical cues such Priscilla M. Wehi, Landcare Research, Private as cuticular hydrocarbons (CHCs) are frequently used by insects to identify suitable Bag 1930 Dunedin, 9054 New Zealand. Email: [email protected] sexual partners. We predicted that New Zealand tree weta (Hemideina spp.), a genus Funding information of nocturnal ensiferan Orthoptera that live both allopatrically and sympatrically, use This work was supported by New Zealand chemical signals from either frass or CHCs to find mates. In a series of six laboratory Foundation for Research, Science and Technology contract number UOWX0501, trials using both H. thoracica and H. crassidens, we found that male tree weta, but not and Postdoctoral fellowship MAUX0905; female tree weta, occupied cavities primed with female cuticular cues more often than Rutherford Discovery Fellowship 14- LCR- 001 to PMW; and Massey University MURF cavities without. However, males did not discriminate between chemical cues of male funding “What limits a weta?” to MMR. and female conspecifics, or between conspecifics and heterospecifics. -

Factors Influencing Occupancy of Modified Artificial Refuges for Monitoring the Range-Restricted Banks Peninsula Tree Weta Hemideina Ricta (Anostostomatidae)
132 AvailableNew on-lineZealand at: Journal http://www.newzealandecology.org/nzje/ of Ecology, Vol. 38, No. 1, 2014 Factors influencing occupancy of modified artificial refuges for monitoring the range-restricted Banks Peninsula tree weta Hemideina ricta (Anostostomatidae) Mike H. Bowie1*, Warwick J. Allen1, Jill McCaw1 and Rachel van Heugten2 1Ecology Department, Agriculture and Life Sciences Division, PO Box 85084, Lincoln University, Lincoln 7647, New Zealand 2School of Biological Sciences, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand *Author for correspondence (Email: [email protected]) Published online: 7 November 2013 Abstract: The use of non-destructive and non-invasive monitoring methods is often necessary for species of high conservation status. Developing monitoring methods to maximise numbers of individuals found is important, given that rare species can be difficult to locate. Artificial refuges called ‘weta motels’ have been used for monitoring tree weta (Orthoptera: Anostostomatidae) since 1992, but poor occupancy for Hemideina ricta and H. femorata necessitated an improved design and assessment of placement to encourage tree weta use. Modification to a basic design of weta motel was tested on New Zealand’s rarest tree weta, H. ricta, on Banks Peninsula, Canterbury, New Zealand. Possible lures such as peanut butter or frass from male and female H. ricta were placed in motels in an attempt to improve occupancy. We recorded high occupancy rates with an improved weta motel design and found that motels containing female frass had significantly higher levels of occupancy than controls, with the former reaching 80% occupation after 6 months. Weta motels were more likely to be used by tree weta in areas with low subcanopy density and patchy or little canopy cover, with H. -

Macroinvertebrate Community Responses to Mammal Control
MACROINVERTEBRATE COMMUNITY RESPONSES TO MAMMAL CONTROL – EVIDENCE FOR TOP-DOWN TROPHIC EFFECTS BY OLIVIA EDITH VERGARA PARRA A thesis submitted to the Victoria University of Wellington in fulfilment of the requirements for the degree of Doctor of Philosophy in Conservation Biology Victoria University of Wellington 2018 Para mi sobrina Violeta Orellana Vergara y su sonrisa hermosa. Tu llegada remeció mi corazón de amor de una manera inimaginable. ¡Sueña en grande! ii Nothing in nature stands alone... (John Hunter 1786) iii iv ABSTRACT New Zealand’s invertebrates are characterised by extraordinary levels of endemism and a tendency toward gigantism, flightlessness and longevity. These characteristics have resulted in a high vulnerability to introduced mammals (i.e. possums, rats, mice, and stoats) which are not only a serious threat to these invertebrates, but have also altered food web interactions over the past two-hundred years. The establishment of fenced reserves and the aerial application of 1080 toxin are two methods of mammal control used in New Zealand to exclude and reduce introduced mammals, respectively. Responses of ground-dwelling invertebrates to mammal control, including a consideration of trophic cascades and their interactions, remain unclear. However, in this thesis, I aimed to investigate how changes in mammal communities inside and outside a fenced reserve (ZEALANDIA, Wellington) and before-and-after the application of 1080 in Aorangi Forest, influence the taxonomic and trophic abundance, body size and other traits of ground-dwelling invertebrates on the mainland of New Zealand. I also tested for effects of habitat variables (i.e. vegetation and elevation), fluctuations in predator populations (i.e. -

Orthoptera: Ensifera)?
Zootaxa 4291 (1): 001–033 ISSN 1175-5326 (print edition) http://www.mapress.com/j/zt/ Article ZOOTAXA Copyright © 2017 Magnolia Press ISSN 1175-5334 (online edition) https://doi.org/10.11646/zootaxa.4291.1.1 http://zoobank.org/urn:lsid:zoobank.org:pub:BD31B828-E7EF-46AD-B618-1BAAA2D63DBD Tackling an intractable problem: Can greater taxon sampling help resolve relationships within the Stenopelmatoidea (Orthoptera: Ensifera)? AMY G. VANDERGAST1,7, DAVID B. WEISSMAN2, DUSTIN A. WOOD3, DAVID C. F. RENTZ4, CORINNA S. BAZELET5 & NORIHIRO UESHIMA6 1U.S. Geological Survey, Western Ecological Research Center, San Diego Field Station, 4165 Spruance Road Suite 200, San Diego, CA 92101, USA. E-mail: [email protected] 2Department of Entomology, California Academy of Sciences, 55 Music Concourse Drive, San Francisco, CA 94118, USA. E-mail: [email protected] 3U.S. Geological Survey, Western Ecological Research Center, San Diego Field Station, 4165 Spruance Road Suite 200, San Diego, CA 92101, USA. E-mail: [email protected] 4School of Marine & Tropical Biology, James Cook University, Australia. E-mail: [email protected] 5Steinhardt Museum, Tel Aviv University, Department of Zoology, Sherman Building Rm. 403, Tel Aviv, Israel; Department of Conser- vation Ecology and Entomology, Stellenbosch University, Private Bag X1, Matieland 7602, South Africa. E-mail: [email protected] 61435-1 Kubocho, Matsusaka, Mie 515-0044, Japan. E-mail: [email protected] 7Corresponding Author Abstract The relationships among and within the families that comprise the orthopteran superfamily Stenopelmatoidea (suborder Ensifera) remain poorly understood. We developed a phylogenetic hypothesis based on Bayesian analysis of two nuclear ribosomal and one mitochondrial gene for 118 individuals (84 de novo and 34 from GenBank). -
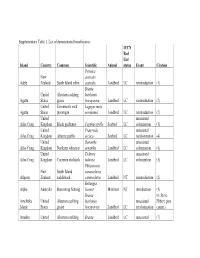
Supplementary Table 1. List of Demonstrated Beneficiaries
Supplementary Table 1. List of demonstrated beneficiaries. IUCN Red List Island Country Common Scientific Animal status Event Citation Petroica New australis Adele Zealand South Island robin australis Landbird LC reintroduction (1) Branta United Aleutian cackling hutchinsii Agattu States goose leucopareia Landbird LC reintroduction (2) United Evermann's rock Lagopus muta Agattu States ptarmigan evermanni Landbird LC reintroduction (2) United unassisted Ailsa Craig Kingdom Black guillemot Cepphus grylle Seabird LC colonization (3) United Fratercula unassisted Ailsa Craig Kingdom Atlantic puffin arctica Seabird LC recolonization (4) United Oenanthe unassisted Ailsa Craig Kingdom Northern wheatear oenanthe Landbird LC colonization (4) United Tadorna unassisted Ailsa Craig Kingdom Common shelduck tadorna Landbird LC colonization (3) Philesturnus New South Island carunculatus Allports Zealand saddleback carunculatus Landbird NT reintroduction (2) Bettongia Alpha Australia Burrowing bettong lesueur Mammal NT introduction (5) Branta (6; Steve Amchitka United Aleutian cackling hutchinsii unassisted Ebbert, pers. Island States goose leucopareia Landbird LC recolonization comm.) Amukta United Aleutian cackling Branta Landbird LC unassisted (7) IUCN Red List Island Country Common Scientific Animal status Event Citation States goose hutchinsii recolonization leucopareia Sally Amy Poncet, Island/Outer United Cinclodes unassisted unpublished Knob Kingdom Tussacbird antarcticus Landbird LC recolonization data Sally Amy Poncet, Island/Outer United unpublished -

Sex- and Season-Dependent Behaviour in a Flightless Insect, the Auckland Tree Weta (Hemideina Thoracica)
AvailableWehi et al.: on-line Seasonal at: http://www.newzealandecology.org/nzje/ behaviour patterns in weta 75 Sex- and season-dependent behaviour in a flightless insect, the Auckland tree weta (Hemideina thoracica) Priscilla M Wehi1,4*, Murray Jorgensen2 and Mary Morgan-Richards3 1Department of Biological Sciences, University of Waikato, Private Bag 3105, Hamilton 3240, New Zealand 2Department of Statistics, University of Waikato, Private Bag 3105, Hamilton 3240, New Zealand 3Institute of Natural Resources, Massey University, Private Bag 11222, Palmerston North, New Zealand 4Present address: CSAFE, University of Otago, P O Box 56, Dunedin, New Zealand *Author for correspondence (Email: [email protected]) Published online: 17 December 2012 Abstract: In a polygynous mating system, males frequently compete by locating and defending sites with resources essential to female survival and reproduction. We investigated seasonal changes in site occupancy in a sexually dimorphic, harem-forming insect, the Auckland tree weta (Hemideina thoracica). First we established artificial cavities as diurnal refuge cavities and potential harem guarding sites. We then examined cavity occupancy changes, and, based on our knowledge of prior occupants, determined sex-specific patterns of arrival, departure, and aggregation at a population level throughout the year. Both season and the sex of prior occupants influenced weta occupancy patterns. Most observations were of single females. However, both males and females moved into cavities previously occupied by a weta of the opposite sex more often than expected by chance alone. Females avoided cavities where other females were present, except during summer when most harems formed. In early summer, male and female tree weta previously living apart began co-habiting. -

Diet Selectivity in a Terrestrial Forest Invertebrate, the Auckland Tree Wētā, Across Three Habitat Zones
CORE Metadata, citation and similar papers at core.ac.uk Provided by Research Commons@Waikato Received: 11 September 2017 | Accepted: 4 December 2017 DOI: 10.1002/ece3.3763 ORIGINAL RESEARCH Diet selectivity in a terrestrial forest invertebrate, the Auckland tree wētā, across three habitat zones Matthew B. G. J. Brown1 | Chrissen E. C. Gemmill1 | Steven Miller2 | Priscilla M. Wehi3 1School of Science, University of Waikato, Hamilton, New Zealand Abstract 2Department of Mathematics and Insects are important but overlooked components of forest ecosystems in New Statistics, School of Computing and Zealand. For many insect species, information on foraging patterns and trophic rela- Mathematical Sciences, University of Waikato, Hamilton, New Zealand tionships is lacking. We examined diet composition and selectivity in a large- bodied 3Centre for Sustainability, University of Otago insect, the Auckland tree wētā Hemideina thoracica, in three habitat zones in a lowland and Manaaki Whenua Landcare Research, New Zealand forest. We asked whether H. thoracica selectively forage from available Dunedin, New Zealand plant food sources, and whether these choices were lipid- rich compared to nonpre- Correspondence ferred available plants. We also identified the proportion of invertebrates in their frass Priscilla M. Wehi, Manaaki Whenua Landcare Research, Dunedin, New Zealand. as a proxy for omnivory. From reconnaissance plot sampling, together with fecal frag- Email: [email protected] ment analysis, we report that more than 93% of individual tree wētā had eaten inver- Funding information tebrates before capture. Additionally, wētā in the highest elevation hillslope habitat University of Waikato; Royal Society of New zone consumed significantly fewer species of plants on average than wētā on the low- Zealand, Grant/Award Number: 14-LCR-001 elevation terrace habitat. -

Weta Affairs: an Investigation Into The
WETA AFFAIRS: AN INVESTIGATION INTO THE POPULATION STRUCTURE AND POSSIBLE HYBRIDISATION OF TWO TREE WETA SPECIES (HEMIDEINA) IN CANTERBURY A thesis submitted in partial fulfilment of the requirements for the Degree of Doctor of Philosophy in Evolutionary Ecology in the University of Canterbury by R. A. van Heugten University of Canterbury 2015 CONTENTS ACKNOWLEDGEMENTS 1 ABSTRACT 2 INTRODUCTION 3 1.1 Genetics and conservation 3 1.1.1 Hybridisation 3 1.1.2 Population Genetics 5 1.1.2.1 Small Populations 5 1.1.2.2 Population structure and landscape genetics 8 1.2 The current study 10 1.2.1 The aims of the current study 15 CHAPTER TWO – Species distribution modelling 17 2.1 Abstract 17 2.2 Introduction 17 2.3 Aim 24 2.4 Method 24 2.4.1 Data 24 2.4.2 Interpolation methods 26 2.4.2.1 Model 1 – Inverse Distance Weight 26 2.4.2.2 Model 2 – Spline with barrier 27 2.4.2.3 Model 3 – Kriging 27 2.4.2.3.1 Model 3A 28 2.4.2.3.2 Model 3B 28 2.4.3 Habitat model 28 2.5 Results 29 2.5.1 Interpolation models 29 2.5.2 Habitat model 35 2.6 Discussion 36 2.7 Conclusion 40 CHAPTER THREE – DNA sampling, species distributions and 41 cohabitation observations 3.1 Abstract 41 3.2 Introduction 41 3.3 Aim 45 3.4 Method 45 3.4.1 Motel design 45 3.4.2 Site locations 46 3.4.2.1 Established motels 46 3.4.2.2 New motels 49 3.4.3 Checking motels 51 3.4.4 Active searching and additional methods 52 3.4.5 DNA Sampling 52 3.4.6 Distribution analysis 55 3.5 Results 55 3.6 Discussion 77 3.7 Conclusion 85 CHAPTER FOUR – Hybridisation 86 4.1 Abstract 86 4.2 Introduction 86 4.3 -

Measuring the Restoration Trajectory of Invertebrates in the Winstone Aggregates Hunua Quarry Site
ISSN: 1177-6242 (Print) ISSN: 1179-7738 (Online) ISBN: 978-0-86476-395-2 (Print) ISBN: 978-0-86476-396-9 (Online) Lincoln University Wildlife Management Report No. 56 Measuring the restoration trajectory of invertebrates in the Winstone Aggregates Hunua quarry site by Erica Stokvis1, Mike Bowie1, Keith Barber2, Cor Vink3 & Bruce Marshall4 1Department of Ecology, Lincoln University, PO Box 85084, Lincoln 7647 2Winstone Aggregates, Hunua Gorge Road, Red Hill 2110, Auckland 3Canterbury Museum, Rolleston Ave, Christchurch 8013 4Museum of New Zealand Te Papa Tongarewa [email protected] [email protected] [email protected] [email protected] [email protected] Prepared for: Winstone Aggregates May 2015 Table of Contents Abstract ...................................................................................................................................... 2 Introduction ............................................................................................................................... 2 Methodology .............................................................................................................................. 3 Results ........................................................................................................................................ 5 Discussion................................................................................................................................. 20 Conclusion ............................................................................................................................... -

Legal Protection of New Zealand's Indigenous Terrestrial Fauna
Tuhinga 25: 25–101 Copyright © Museum of New Zealand Te Papa Tongarewa (2014) Legal protection of New Zealand’s indigenous terrestrial fauna – an historical review Colin M. Miskelly Museum of New Zealand Te Papa Tongarewa, PO Box 467, Wellington, New Zealand ([email protected]) ABSTRACT: New Zealand has had a complex history of wildlife protection, with at least 609 different pieces of legislation affecting the protection of native wildlife between 1861 and 2013. The first species to be fully protected was the tüï (Prosthemadera novaeseelandiae), which was listed as a native game species in 1873 and excluded from hunting in all game season notices continuously from 1878, until being absolutely protected in 1906. The white heron (Ardea modesta) and crested grebe (Podiceps cristatus) were similarly protected nationwide from 1888, and the huia (Heteralocha acutirostris) from 1892. Other species listed as native game before 1903 were not consistently excluded from hunting in game season notices, meaning that such iconic species as kiwi (Apteryx spp.), käkäpö (Strigops habroptilus), kökako (Callaeas spp.), saddlebacks (Philesturnus spp.), stitchbird (Notiomystis cincta) and bellbird (Anthornis melanura) could still be taken or killed during the game season until they were absolutely protected in 1906. The tuatara (Sphenodon punctatus) was added to the native game list in 1895, but due to inadequate legislation was not absolutely protected until 1907. The Governor of the Colony of New Zealand had the power to absolutely protect native birds from 1886, but this was not used until 1903, when first the blue duck (Hymenolaimus malacorhynchus) and then the huia were given the status of absolutely protected, followed by more than 130 bird species by the end of 1906.